 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3has: Crystal structure of bacteriorhodopsin mutant L152A crystallized ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3has | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of bacteriorhodopsin mutant L152A crystallized from bicelles | ||||||
 Components Components | Bacteriorhodopsin | ||||||
 Keywords Keywords | TRANSPORT PROTEIN / bacteriorhodopsin / packing force / van der Waals / evolutionary constraint / membrane protein / integral membrane protein / helical membrane protein / proton transport / Cell membrane / Chromophore / Hydrogen ion transport / Ion transport / Membrane / Photoreceptor protein / Pyrrolidone carboxylic acid / Receptor / Retinal protein / Sensory transduction / Transmembrane / Transport | ||||||
| Function / homology |  Function and homology information Function and homology informationlight-driven active monoatomic ion transmembrane transporter activity / monoatomic ion channel activity / photoreceptor activity / phototransduction / proton transmembrane transport / plasma membrane Similarity search - Function | ||||||
| Biological species |  Halobacterium salinarum (Halophile) Halobacterium salinarum (Halophile) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å | ||||||
 Authors Authors | Joh, N.H. / Yang, D. / Bowie, J.U. | ||||||
 Citation Citation | Journal: J.Am.Chem.Soc. / Year: 2009 Title: Similar energetic contributions of packing in the core of membrane and water-soluble proteins. Authors: Joh, N.H. / Oberai, A. / Yang, D. / Whitelegge, J.P. / Bowie, J.U. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3has.cif.gz | 66.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3has.ent.gz | 47.1 KB | Display | PDB format |
| PDBx/mmJSON format | 3has.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 3has_validation.pdf.gz | 888 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 3has_full_validation.pdf.gz | 899.6 KB | Display | |
| Data in XML | 3has_validation.xml.gz | 13.8 KB | Display | |
| Data in CIF | 3has_validation.cif.gz | 18.3 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ha/3hasftp://data.pdbj.org/pub/pdb/validation_reports/ha/3has | HTTPS FTP |
-Related structure data















-Links
 PDBj
PDBj







- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 26887.420 Da / Num. of mol.: 1 / Mutation: L152A Source method: isolated from a genetically manipulated source Source: (gene. exp.) Halobacterium salinarum (Halophile) / Gene: bop, VNG_1467G / Production host: Halobacterium salinarum (Halophile) / Strain (production host): L33 / References: UniProt: P02945 |
|---|
-Non-polymers , 8 types, 90 molecules 


| #2: Chemical | ChemComp-RET /  Mass: 284.436 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H28O Mass: 284.436 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H28O | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
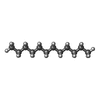
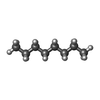
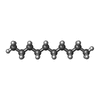
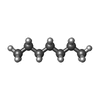
| #3: Chemical |  Mass: 170.335 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C12H26 Mass: 170.335 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C12H26#4: Chemical | ChemComp-OCT / |  Mass: 114.229 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18 Mass: 114.229 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18#5: Chemical |  Mass: 142.282 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H22 Mass: 142.282 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H22#6: Chemical | ChemComp-R16 / |  Mass: 226.441 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H34 Mass: 226.441 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H34#7: Chemical | ChemComp-HP6 / |  Mass: 100.202 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H16 Mass: 100.202 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H16#8: Chemical | ChemComp-CPS / |  Mass: 614.877 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C32H58N2O7S / Comment: detergent*YM Mass: 614.877 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C32H58N2O7S / Comment: detergent*YM#9: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 80 / Source method: isolated from a natural source / Formula: H2O |
-Details
| Has protein modification | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.76 Å3/Da / Density % sol: 55.38 % |
|---|---|
| Crystal grow | Temperature: 310 K / Method: hanging drop, bicelle method / pH: 4 Details: 1.68 M sodium phosphate, 180 mM 1,6-hexanediol, 3.5 % triethylene glycol, PFPC used as cryoprotectant, pH 4.0, hanging drop, bicelle method, temperature 310K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS  / Beamline: 8.2.1 / Wavelength: 1 Å / Beamline: 8.2.1 / Wavelength: 1 Å |
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Apr 13, 2008 |
| Radiation | Monochromator: KOHZU / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→90 Å / Num. obs: 23760 / % possible obs: 99.2 % / Redundancy: 7.4 % / Rmerge(I) obs: 0.126 / Χ2: 1.006 / Net I/σ(I): 13.945 |
| Reflection shell | Resolution: 1.9→1.97 Å / Redundancy: 2.9 % / Rmerge(I) obs: 0.413 / Num. unique all: 2192 / Χ2: 1.011 / % possible all: 93 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.9→27.23 Å / Cor.coef. Fo:Fc: 0.955 / Cor.coef. Fo:Fc free: 0.942 / Occupancy max: 1 / Occupancy min: 0.2 / SU B: 5.322 / SU ML: 0.073 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.131 / ESU R Free: 0.122 Stereochemistry target values: MAXIMUM LIKELIHOOD WITH PHASES Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 68.36 Å2 / Biso mean: 24.964 Å2 / Biso min: 13.18 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→27.23 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.9→1.949 Å / Total num. of bins used: 20
|
