 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3fnv: Crystal Structure of Miner1: The Redox-active 2Fe-2S Protein Caus... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3fnv | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of Miner1: The Redox-active 2Fe-2S Protein Causative in Wolfram Syndrome 2 | ||||||
 Components Components | CDGSH iron sulfur domain-containing protein 2 | ||||||
 Keywords Keywords | METAL BINDING PROTEIN / diabetes / membrane bound / thiazolidinedione / oxidative stress / CDGSH / Endoplasmic reticulum / Iron / Iron-sulfur / Membrane / Metal-binding / Transmembrane / Zinc-finger | ||||||
| Function / homology |  Function and homology information Function and homology informationperinuclear endoplasmic reticulum / autophagy of mitochondrion / regulation of autophagy / 2 iron, 2 sulfur cluster binding / mitochondrial outer membrane / endoplasmic reticulum membrane / endoplasmic reticulum / protein homodimerization activity / protein-containing complex / RNA binding ...perinuclear endoplasmic reticulum / autophagy of mitochondrion / regulation of autophagy / 2 iron, 2 sulfur cluster binding / mitochondrial outer membrane / endoplasmic reticulum membrane / endoplasmic reticulum / protein homodimerization activity / protein-containing complex / RNA binding / membrane / metal ion binding Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.1 Å X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.1 Å | ||||||
 Authors Authors | Conlan, A.R. / Axelrod, H.L. / Cohen, A.E. / Abresch, E.C. / Yee, D. / Zuris, J. / Nechushtai, R. / Jennings, P.A. / Paddock, M.L. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2009 Title: Crystal structure of Miner1: The redox-active 2Fe-2S protein causative in Wolfram Syndrome 2. Authors: Conlan, A.R. / Axelrod, H.L. / Cohen, A.E. / Abresch, E.C. / Zuris, J. / Yee, D. / Nechushtai, R. / Jennings, P.A. / Paddock, M.L. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3fnv.cif.gz | 42.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3fnv.ent.gz | 28.7 KB | Display | PDB format |
| PDBx/mmJSON format | 3fnv.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/fn/3fnvftp://data.pdbj.org/pub/pdb/validation_reports/fn/3fnv | HTTPS FTP |
|---|
-Related structure data
| Related structure data | |
|---|---|
| Similar structure data |



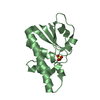







-Links
 PDBj
PDBj







- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||||||||
| Unit cell |
| ||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Component-ID: 1 / Ens-ID: 1 / Refine code: 4 / Auth seq-ID: 68 - 500 / Label seq-ID: 68 - 500
| ||||||||||||||||||
| Details | The biological assembly is a dimer formed from the two chains (A and B) in the crystallographic asymmetric unit. |
-Components
| #1: Protein | Mass: 9490.047 Da / Num. of mol.: 2 Fragment: C-terminal water-soluble domain: UNP residues 57-135 Mutation: C92S Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: CDGSH2, CISD2, ERIS, ZCD2 / Plasmid: pET28a(+) / Production host:  #2: Chemical | 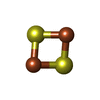  Mass: 175.820 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Fe2S2 Mass: 175.820 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Fe2S2#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 40 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 40 / Source method: isolated from a natural source / Formula: H2OSequence details | 1. IN THE TARGET SEQUENCE, CYS 92 IS REPLACED BY AN SER RESIDUE BY SITE-DIRECTED MUTAGENESIS. 2. ...1. IN THE TARGET SEQUENCE, CYS 92 IS REPLACED BY AN SER RESIDUE BY SITE-DIRECTED MUTAGENESI | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.94 Å3/Da / Density % sol: 36.58 % |
|---|---|
| Crystal grow | Temperature: 300 K / Method: vapor diffusion / pH: 8 Details: 100 mM Tris-HCl pH 8.0, 100 mM NaCl, 15% PEG 3000, VAPOR DIFFUSION |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL  / Beamline: BL9-2 / Wavelength: 1.7418, 1.3624, 1.7372 / Beamline: BL9-2 / Wavelength: 1.7418, 1.3624, 1.7372 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: MARMOSAIC 325 mm CCD / Detector: CCD / Date: Dec 15, 2008 / Details: Flat collimating mirror, toroid focusing mirror | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: Double crystal / Protocol: MAD / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Number: 217494 / Rmerge(I) obs: 0.139 / D res high: 2.16 Å / Num. obs: 8210 / % possible obs: 97.9 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Diffraction reflection shell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.8→48.56 Å / Num. obs: 14086 / % possible obs: 98.6 % / Observed criterion σ(I): -3 / Redundancy: 25.9 % / Biso Wilson estimate: 34.594 Å2 / Rmerge(I) obs: 0.143 / Net I/σ(I): 16.17 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Resolution: 1.8→1.86 Å / Redundancy: 17.9 % / Rmerge(I) obs: 1.79 / Mean I/σ(I) obs: 1.9 / Num. measured obs: 18126 / Num. unique obs: 1285 / % possible all: 97.9 |
-Phasing
| Phasing | Method: MAD | |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Phasing MAD set site |
| |||||||||||||||||||||||||||||||||||||||||||||||||
| Phasing dm | FOM : 0.8 / FOM acentric: 0.81 / FOM centric: 0.74 / Reflection: 9018 / Reflection acentric: 7562 / Reflection centric: 1456 | |||||||||||||||||||||||||||||||||||||||||||||||||
| Phasing dm shell |
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 2.1→37.06 Å / Cor.coef. Fo:Fc: 0.96 / Cor.coef. Fo:Fc free: 0.943 / WRfactor Rfree: 0.235 / WRfactor Rwork: 0.199 / Occupancy max: 1 / Occupancy min: 0.5 / FOM work R set: 0.928 / SU B: 7.676 / SU ML: 0.101 / SU R Cruickshank DPI: 0.208 / SU Rfree: 0.172 / TLS residual ADP flag: LIKELY RESIDUAL / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.183 / ESU R Free: 0.164 Stereochemistry target values: MAXIMUM LIKELIHOOD WITH PHASES Details: 1. HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. 2. ATOM RECORD CONTAINS RESIDUAL B FACTORS ONLY. 3. AN 2FE-2S CLUSTER (FES) WAS MODELED INTO EACH SUBUNIT IN THE ASYMMETRIC UNIT. THE ...Details: 1. HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. 2. ATOM RECORD CONTAINS RESIDUAL B FACTORS ONLY. 3. AN 2FE-2S CLUSTER (FES) WAS MODELED INTO EACH SUBUNIT IN THE ASYMMETRIC UNIT. THE PRESENCE OF THE 2FE-2S CLUSTER WAS CORROBORATED BY ANOMALOUS DIFFERENCE MAPS. THE PROTEIN LIGANDS TO THE FE ATOMS IN THE 2FE-2S CLUSTERS ARE CYS 99, CYS 101, CYS 110, AND HIS 114.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 59.35 Å2 / Biso mean: 35.795 Å2 / Biso min: 16.62 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.1→37.06 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Dom-ID: 1 / Auth asym-ID: A / Ens-ID: 1 / Number: 775 / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.1→2.154 Å / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
