 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3fhj: Independent saturation of three TrpRS subsites generates a partia... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3fhj | ||||||
|---|---|---|---|---|---|---|---|
| Title | Independent saturation of three TrpRS subsites generates a partially-assembled state similar to those observed in molecular simulations | ||||||
 Components Components | Tryptophanyl-tRNA synthetase | ||||||
 Keywords Keywords | TRANSLATION / ligand-dependent domain rearrangement / mechanistic pathway / molecular simulations / Aminoacyl-tRNA synthetase / ATP-binding / Cytoplasm / Ligase / Nucleotide-binding / Protein biosynthesis | ||||||
| Function / homology |  Function and homology information Function and homology informationtryptophan-tRNA ligase / tryptophanyl-tRNA aminoacylation / tryptophan-tRNA ligase activity / ATP binding / cytosol Similarity search - Function | ||||||
| Biological species |   Bacillus stearothermophilus (bacteria) Bacillus stearothermophilus (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.65 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.65 Å | ||||||
 Authors Authors | Laowanapiban, P. / Kapustina, M. / Vonrhein, C. / Delarue, M. / Koehl, P. / Carter Jr., C.W. | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.Usa / Year: 2009 Title: Independent saturation of three TrpRS subsites generates a partially assembled state similar to those observed in molecular simulations. Authors: Laowanapiban, P. / Kapustina, M. / Vonrhein, C. / Delarue, M. / Koehl, P. / Carter, C.W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3fhj.cif.gz | 352.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3fhj.ent.gz | 287.9 KB | Display | PDB format |
| PDBx/mmJSON format | 3fhj.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/fh/3fhjftp://data.pdbj.org/pub/pdb/validation_reports/fh/3fhj | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3fi0SC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |
























-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 37225.672 Da / Num. of mol.: 6 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Bacillus stearothermophilus (bacteria) / Strain: NC1500 / Gene: trpS / Plasmid: PET42 / Production host: #2: Chemical | ChemComp-TRP / 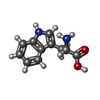  Type: L-peptide linking / Mass: 204.225 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C11H12N2O2 Type: L-peptide linking / Mass: 204.225 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C11H12N2O2#3: Chemical | ChemComp-PO4 /   Mass: 94.971 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: PO4 Mass: 94.971 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: PO4#4: Chemical | ChemComp-AMP /   Mass: 347.221 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C10H14N5O7P / Comment: AMP*YM Mass: 347.221 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C10H14N5O7P / Comment: AMP*YM |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.63 Å3/Da / Density % sol: 53.22 % |
|---|---|
| Crystal grow | Temperature: 315 K / pH: 6.58 Details: 2.28M K2HPO4, 0.6% (v/v) PEG 400, pH 6.58, dialysis, temperature 315K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 22-ID / Wavelength: 1 / Beamline: 22-ID / Wavelength: 1 |
| Detector | Type: MAR scanner 300 mm plate / Detector: IMAGE PLATE / Date: Sep 1, 2006 |
| Radiation | Monochromator: APS SERCAT 22ID BEAMLINE / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.5→25 Å / Num. obs: 161648 / % possible obs: 97.6 % / Observed criterion σ(I): 3 / Redundancy: 3.48 % / Biso Wilson estimate: 52.66 Å2 / Rmerge(I) obs: 0.081 |
| Reflection shell | Resolution: 2.65→2.85 Å / Redundancy: 5.9 % / Rmerge(I) obs: 0.414 / Mean I/σ(I) obs: 4.22 / % possible all: 77.5 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3FI0 Resolution: 2.65→25 Å / Occupancy max: 1 / Occupancy min: 0.33 Isotropic thermal model: We alternated the use of TLS refinement in REFMAC5 and mapping these parameters to individual isotropic B-values, which were fixed in coordinate refinements with Buster 2.0. Cross valid method: THROUGHOUT / σ(F): 1.5 / Stereochemistry target values: ENGH & HUBER
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 63.5 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.65→25 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.5→2.56 Å
|
