 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3fg5: Crystal structure determination of a ternary complex of phospholi... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3fg5 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure determination of a ternary complex of phospholipase A2 with a pentapeptide FLSYK and Ajmaline at 2.5 A resolution | ||||||
 Components Components |
| ||||||
 Keywords Keywords | HYDROLASE / pla2 / pentapeptide / FLSYK / ajmaline / ternary complex | ||||||
| Function / homology | Phospholipase A2 / Phospholipase A2 domain / Up-down Bundle / Mainly Alpha / AJMALINE Function and homology information Function and homology information | ||||||
| Biological species |  Daboia russelli pulchella (snake) Daboia russelli pulchella (snake) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.5 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.5 Å | ||||||
 Authors Authors | Kumar, M. / Kumar, S. / Vikram, G. / Singh, N. / Sinha, M. / Bhushan, A. / Kaur, P. / Srinivasan, A. / Sharma, S. / Singh, T.P. | ||||||
 Citation Citation | Journal: To be Published Title: Crystal structure determination of a ternary complex of phospholipase A2 with a pentapeptide FLSYK and Ajmaline at 2.5 A resolution Authors: Kumar, M. / Kumar, S. / Vikram, G. / Singh, N. / Sinha, M. / Bhushan, A. / Kaur, P. / Srinivasan, A. / Sharma, S. / Singh, T.P. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3fg5.cif.gz | 39.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3fg5.ent.gz | 27 KB | Display | PDB format |
| PDBx/mmJSON format | 3fg5.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 3fg5_validation.pdf.gz | 695.6 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 3fg5_full_validation.pdf.gz | 700.5 KB | Display | |
| Data in XML | 3fg5_validation.xml.gz | 9 KB | Display | |
| Data in CIF | 3fg5_validation.cif.gz | 11.2 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/fg/3fg5ftp://data.pdbj.org/pub/pdb/validation_reports/fg/3fg5 | HTTPS FTP |
-Related structure data
| Related structure data |  1zr8S S: Starting model for refinement |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 13629.767 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Daboia russelli pulchella (snake) / References: phospholipase A2 |
|---|---|
| #2: Protein/peptide | Mass: 657.778 Da / Num. of mol.: 1 / Source method: obtained synthetically / Details: SYNTHESIZED SHORT PEPTIDE |
| #3: Chemical | ChemComp-AJM / 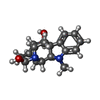  Mass: 298.379 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H22N2O2 Mass: 298.379 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H22N2O2 |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 61 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 61 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
| Sequence details | THE SEQUENCE OF ENTITY 1 IS THE SAME WITH UNP P59071 PA28_DABRP, WHICH HAS DIFFERENT SOURCE. |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.38 Å3/Da / Density % sol: 48.24 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 6.8 Details: 0.2M AMMONIUM SULPHATE, 0.2M AMMONIUM ACETATE, 30% PEG 4000, pH 6.8, VAPOR DIFFUSION, HANGING DROP, temperature 298K |
-Data collection
| Diffraction | Mean temperature: 300 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU300 / Wavelength: 1.5418 Å |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Nov 2, 2007 / Details: Mirror |
| Radiation | Monochromator: Graphite / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.5→20 Å / Num. all: 4639 / Num. obs: 4392 / % possible obs: 93.2 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Rsym value: 0.096 / Net I/σ(I): 6.4 |
| Reflection shell | Resolution: 2.5→2.59 Å / Mean I/σ(I) obs: 1.7 / Rsym value: 0.439 / % possible all: 98.5 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1ZR8 Resolution: 2.5→20 Å / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / Stereochemistry target values: MAXIMUM LIKELIHOOD
| |||||||||||||||||||||||||
| Displacement parameters | Biso max: 98.14 Å2 / Biso min: 11.91 Å2
| |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.5→20 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
|
