 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3cgt: STRUCTURE OF CYCLODEXTRIN GLYCOSYLTRANSFERASE COMPLEXED WITH ITS ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3cgt | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | STRUCTURE OF CYCLODEXTRIN GLYCOSYLTRANSFERASE COMPLEXED WITH ITS MAIN PRODUCT BETA-CYCLODEXTRIN | |||||||||
 Components Components | CYCLODEXTRIN GLYCOSYLTRANSFERASE | |||||||||
 Keywords Keywords | GLYCOSYLTRANSFERASE / STARCH DEGRADATION / CYCLODEXTRIN | |||||||||
| Function / homology |  Function and homology information Function and homology informationcyclomaltodextrin glucanotransferase / cyclomaltodextrin glucanotransferase activity / starch binding / alpha-amylase activity / carbohydrate metabolic process / extracellular region / metal ion binding Similarity search - Function | |||||||||
| Biological species |  Bacillus circulans (bacteria) Bacillus circulans (bacteria) | |||||||||
| Method |  X-RAY DIFFRACTION / DIFFERENCE FOURIER / Resolution: 2.4 Å X-RAY DIFFRACTION / DIFFERENCE FOURIER / Resolution: 2.4 Å | |||||||||
 Authors Authors | Schmidt, A.K. / Schulz, G.E. | |||||||||
 Citation Citation | Journal: Biochemistry / Year: 1998 Title: Structure of cyclodextrin glycosyltransferase complexed with a derivative of its main product beta-cyclodextrin. Authors: Schmidt, A.K. / Cottaz, S. / Driguez, H. / Schulz, G.E. #1: Journal: Biochemistry / Year: 1992Title: Catalytic Center of Cyclodextrin Glycosyltransferase Derived from X-Ray Structure Analysis Combined with Site-Directed Mutagenesis Authors: Klein, C. / Hollender, J. / Bender, H. / Schulz, G.E. #2: Journal: J.Mol.Biol. / Year: 1991Title: Structure of Cyclodextrin Glycosyltransferase Refined at 2.0 A Resolution Authors: Klein, C. / Schulz, G.E. #3: Journal: Appl.Microbiol.Biotechnol. / Year: 1990Title: Molecular Cloning, Nucleotide Sequence and Expression in Escherichia Coli of the Beta-Cyclodextrin Glycosyltransferase Gene from Bacillus Circulans Strain No. 8 Authors: Nitschke, L. / Heeger, K. / Bender, H. / Schulz, G.E. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3cgt.cif.gz | 148.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3cgt.ent.gz | 115.1 KB | Display | PDB format |
| PDBx/mmJSON format | 3cgt.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/cg/3cgtftp://data.pdbj.org/pub/pdb/validation_reports/cg/3cgt | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 74507.008 Da / Num. of mol.: 1 / Mutation: E257A Source method: isolated from a genetically manipulated source Source: (gene. exp.) Bacillus circulans (bacteria) / Strain: 8 / Cellular location: EXTRACELLULAR / Production host: References: UniProt: P30920, cyclomaltodextrin glucanotransferase | ||||
|---|---|---|---|---|---|
| #2: Polysaccharide | Cycloheptakis-(1-4)-(alpha-D-glucopyranose) / beta-cyclodextrin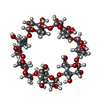  Source method: isolated from a genetically manipulated source Details: cyclic oligosaccharide / References: beta-cyclodextrin | ||||
| #3: Chemical |   Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 205 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 205 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.79 Å3/Da / Density % sol: 67.53 % | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 6.9 / Details: pH 6.9 | ||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 6.7 / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 298 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RUH2R / Wavelength: 1.5418 |
| Detector | Type: SIEMENS / Detector: AREA DETECTOR / Date: May 1, 1996 |
| Radiation | Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.35→29.24 Å / Num. obs: 42278 / % possible obs: 89.1 % / Redundancy: 4.3 % / Rsym value: 0.096 / Net I/σ(I): 7.2 |
| Reflection shell | Resolution: 2.41→2.48 Å / Mean I/σ(I) obs: 2.5 / Rsym value: 0.266 / % possible all: 71.5 |
| Reflection | *PLUS Num. measured all: 183009 / Rmerge(I) obs: 0.096 |
| Reflection shell | *PLUS % possible obs: 71.5 % / Rmerge(I) obs: 0.266 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: DIFFERENCE FOURIER / Resolution: 2.4→30 Å / Cross valid method: THROUGHOUT
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 25.3 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.4→30 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.4→2.51 Å / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.1F / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor obs: 0.288 |
