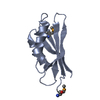
 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3aqe | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the extracellular domain of human RAMP2 | ||||||
 Components Components | Receptor activity-modifying protein 2 | ||||||
 Keywords Keywords | TRANSPORT PROTEIN / Transmembrane / GPCR / Adrenomedullin / TRAFFICKING / CLR / CGRP / Endoplasmic Reticulum / DISEASE / Neovascularization / Helix bundle / Calcitonin Receptor-Like Receptor (CRLR) / Endoplasmic Reticulum (ER) / Cell membrane | ||||||
| Function / homology |  Function and homology information Function and homology informationbasement membrane assembly / adrenomedullin binding / vascular associated smooth muscle cell development / adrenomedullin receptor activity / adrenomedullin receptor complex / adrenomedullin receptor signaling pathway / amylin receptor 2 signaling pathway / positive regulation of vasculogenesis / bicellular tight junction assembly / Calcitonin-like ligand receptors ...basement membrane assembly / adrenomedullin binding / vascular associated smooth muscle cell development / adrenomedullin receptor activity / adrenomedullin receptor complex / adrenomedullin receptor signaling pathway / amylin receptor 2 signaling pathway / positive regulation of vasculogenesis / bicellular tight junction assembly / Calcitonin-like ligand receptors / regulation of G protein-coupled receptor signaling pathway / negative regulation of vascular permeability / sprouting angiogenesis / adherens junction assembly / cellular response to vascular endothelial growth factor stimulus / negative regulation of endothelial cell apoptotic process / vasculogenesis / cellular response to hormone stimulus / coreceptor activity / clathrin-coated pit / protein localization to plasma membrane / intracellular protein transport / receptor internalization / regulation of blood pressure / adenylate cyclase-activating G protein-coupled receptor signaling pathway / positive regulation of angiogenesis / calcium ion transport / protein transport / heart development / High laminar flow shear stress activates signaling by PIEZO1 and PECAM1:CDH5:KDR in endothelial cells / angiogenesis / G alpha (s) signalling events / lysosome / receptor complex / G protein-coupled receptor signaling pathway / positive regulation of gene expression / cell surface / plasma membrane / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2 Å X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2 Å | ||||||
 Authors Authors | Kusano, S. / Kukimoto-Niino, M. / Shirouzu, M. / Shindo, T. / Yokoyama, S. | ||||||
 Citation Citation | Journal: Protein Sci. / Year: 2012 Title: Structural basis for extracellular interactions between calcitonin receptor-like receptor and receptor activity-modifying protein 2 for adrenomedullin-specific binding Authors: Kusano, S. / Kukimoto-Niino, M. / Hino, N. / Ohsawa, N. / Okuda, K. / Sakamoto, K. / Shirouzu, M. / Shindo, T. / Yokoyama, S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3aqe.cif.gz | 111.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3aqe.ent.gz | 88.1 KB | Display | PDB format |
| PDBx/mmJSON format | 3aqe.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 3aqe_validation.pdf.gz | 469.8 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 3aqe_full_validation.pdf.gz | 475.6 KB | Display | |
| Data in XML | 3aqe_validation.xml.gz | 23.9 KB | Display | |
| Data in CIF | 3aqe_validation.cif.gz | 32.6 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/aq/3aqeftp://data.pdbj.org/pub/pdb/validation_reports/aq/3aqe | HTTPS FTP |
-Related structure data









-Links
 PDBj
PDBj- Assembly
Assembly









-Components
| #1: Protein | Mass: 10581.471 Da / Num. of mol.: 6 / Fragment: extracellular Domain, UNP residues 56-139 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: RAMP2 / Plasmid: PX070809-03 / Production host: Escherichia coli cell-free system / References: UniProt: O60895#2: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 282 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 282 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.94 Å3/Da / Density % sol: 36.48 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 7.2 Details: 0.1M Sodium-HEPES, 25% (v/v) PEG 400, 0.2M Calcium chloride di-hydrate, 0.15% Agarose , pH 7.2, VAPOR DIFFUSION, HANGING DROP, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SPring-8  / Beamline: BL26B2 / Wavelength: 0.9788, 0.9792, 0.9640 / Beamline: BL26B2 / Wavelength: 0.9788, 0.9792, 0.9640 | ||||||||||||
| Detector | Type: RIGAKU JUPITER 210 / Detector: CCD / Date: Jun 6, 2008 | ||||||||||||
| Radiation | Monochromator: Si double crystal / Protocol: MAD / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||
| Radiation wavelength |
| ||||||||||||
| Reflection | Resolution: 2→50 Å / Num. obs: 33866 / % possible obs: 100 % / Observed criterion σ(I): -3 / Redundancy: 6.62 % / Rsym value: 0.084 / Net I/σ(I): 19.3 | ||||||||||||
| Reflection shell | Resolution: 2→2.07 Å / Mean I/σ(I) obs: 7.7 / Rsym value: 0.24 / % possible all: 99.1 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 2→36.408 Å / Occupancy max: 1 / Occupancy min: 1 / FOM work R set: 0.7331 / SU ML: 0.37 / Isotropic thermal model: RESTRAINED / σ(F): 1.33 / Phase error: 31.76 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.98 Å / VDW probe radii: 1.2 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 35.941 Å2 / ksol: 0.376 e/Å3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 124.91 Å2 / Biso mean: 28.0343 Å2 / Biso min: 7.88 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→36.408 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 12
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
|
