 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 344d | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| タイトル | DETERMINATION BY MAD-DM OF THE STRUCTURE OF THE DNA DUPLEX D(ACGTACG(5-BRU))2 AT 1.46A AND 100K | ||||||||||||||||||
 要素 要素 | DNA (5'-D(* キーワード キーワードDNA / A-DNA / DOUBLE HELIX / MODIFIED | 機能・相同性 | DNA |  機能・相同性情報 機能・相同性情報手法 |  X線回折 / シンクロトロン / 多波長異常分散 / 解像度: 1.46 Å X線回折 / シンクロトロン / 多波長異常分散 / 解像度: 1.46 Å  データ登録者 データ登録者Todd, A.R. / Adams, A. / Powell, H.R. / Cardin, C.J. |  引用ジャーナル: Acta Crystallogr.,Sect.D / 年: 1999 引用ジャーナル: Acta Crystallogr.,Sect.D / 年: 1999タイトル: Determination by MAD-DM of the structure of the DNA duplex d[ACGTACG(5-BrU)]2 at 1.46 A and 100 K. 著者: Todd, A.K. / Adams, A. / Powell, H.R. / Wilcock, D.J. / Thorpe, J.H. / Lausi, A. / Zanini, F. / Wakelin, L.P. / Cardin, C.J. 履歴 |
|
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 344d.cif.gz | 21.4 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb344d.ent.gz | 15 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 344d.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/44/344dftp://data.pdbj.org/pub/pdb/validation_reports/44/344d | HTTPS FTP |
|---|
-関連構造データ
| 関連構造データ |  243dS S: 精密化の開始モデル |
|---|---|
| 類似構造データ |







-リンク
 PDBj
PDBj

- 集合体
集合体
| 登録構造単位 | 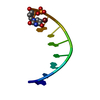
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||
| 単位格子 |
| ||||||||||||
| Components on special symmetry positions |
|
-要素
| #1: DNA鎖 | 分子量: 2491.487 Da / 分子数: 1 / 由来タイプ: 合成 |
|---|---|
| #2: 水 | ChemComp-HOH /  分子量: 18.015 Da / 分子数: 88 / 由来タイプ: 天然 / 式: H2O 分子量: 18.015 Da / 分子数: 88 / 由来タイプ: 天然 / 式: H2O |
-実験情報
-実験
| 実験 | 手法: X線回折 / 使用した結晶の数: 1 |
|---|
- 試料調製
試料調製
| 結晶 | マシュー密度: 2.5 Å3/Da / 溶媒含有率: 50.9 % | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 結晶化 | pH: 6.5 / 詳細: pH 6.50 | |||||||||||||||||||||||||||||||||||
| 結晶化 | *PLUS 温度: 285 K / 手法: 蒸気拡散法, ハンギングドロップ法 | |||||||||||||||||||||||||||||||||||
| 溶液の組成 | *PLUS
|
-データ収集
| 回折 | 平均測定温度: 100 K |
|---|---|
| 放射光源 | 由来: シンクロトロン / サイト: ELETTRA  / ビームライン: 5.2R / ビームライン: 5.2R |
| 検出器 | タイプ: MARRESEARCH / 検出器: IMAGE PLATE / 日付: 1996年5月14日 / 詳細: MIRROR |
| 放射 | モノクロメーター: SI CRYSTAL / プロトコル: SINGLE WAVELENGTH / 単色(M)・ラウエ(L): M / 散乱光タイプ: x-ray |
| 放射波長 | 相対比: 1 |
| 反射 | 解像度: 1.4→15 Å / Num. obs: 8062 / % possible obs: 98.3 % / 冗長度: 4.35 % / Rmerge(I) obs: 0.071 / Net I/σ(I): 0.137 |
| 反射 シェル | 解像度: 1.4→1.45 Å / 冗長度: 1.1 % / Rmerge(I) obs: 0.137 / % possible all: 60.1 |
| 反射 | *PLUS 最高解像度: 1.4 Å / 最低解像度: 15 Å / % possible obs: 98.3 % / 冗長度: 4.35 % |
| 反射 シェル | *PLUS 最高解像度: 1.4 Å / 最低解像度: 1.45 Å / % possible obs: 60.1 % / 冗長度: 1.1 % |
- 解析
解析
| ソフトウェア |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 精密化 | 構造決定の手法: 多波長異常分散 開始モデル: NDB ENTRY ADH070 (PDB ENTRY 243D) 最高解像度: 1.46 Å / Num. parameters: 2232 / Num. restraintsaints: 2730 / σ(F): 0 / 立体化学のターゲット値: ENGH AND HUBER / 詳細: ANISOTROPIC REFINEMENT /
| |||||||||||||||||||||||||||||||||
| 溶媒の処理 | 溶媒モデル: MOEWS & KRETSINGER,J.MOL.BIOL.91(1973)201 | |||||||||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 0 / Occupancy sum hydrogen: 0 / Occupancy sum non hydrogen: 245 | |||||||||||||||||||||||||||||||||
| 精密化ステップ | サイクル: LAST / 最高解像度: 1.46 Å
| |||||||||||||||||||||||||||||||||
| 拘束条件 |
| |||||||||||||||||||||||||||||||||
| ソフトウェア | *PLUS 名称: SHELXL-96 / 分類: refinement | |||||||||||||||||||||||||||||||||
| 精密化 | *PLUS Num. reflection obs: 4312 / σ(F): 0 / Rfactor all: 0.1115 / Rfactor obs: 0.109 / Rfactor Rfree: 0.2003 | |||||||||||||||||||||||||||||||||
| 溶媒の処理 | *PLUS | |||||||||||||||||||||||||||||||||
| 原子変位パラメータ | *PLUS | |||||||||||||||||||||||||||||||||
| 拘束条件 | *PLUS
|
