 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2yvj: Crystal structure of the ferredoxin-ferredoxin reductase (BPHA3-B... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2yvj | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the ferredoxin-ferredoxin reductase (BPHA3-BPHA4)complex | ||||||
 Components Components |
| ||||||
 Keywords Keywords | OXIDOREDUCTASE/ELECTRON TRANSPORT / electron transfer / ferredoxin / ferredoxin reductase / OXIDOREDUCTASE-ELECTRON TRANSPORT COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationoxidoreductase activity, acting on NAD(P)H / 2 iron, 2 sulfur cluster binding / nucleotide binding / metal ion binding / cytoplasm Similarity search - Function | ||||||
| Biological species |  Pseudomonas sp. (bacteria) Pseudomonas sp. (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å | ||||||
 Authors Authors | Senda, T. / Senda, M. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2007 Title: Molecular Mechanism of the Redox-dependent Interaction between NADH-dependent Ferredoxin Reductase and Rieske-type [2Fe-2S] Ferredoxin Authors: Senda, M. / Kishigami, S. / Kimura, S. / Fukuda, M. / Ishida, T. / Senda, T. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2yvj.cif.gz | 188.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2yvj.ent.gz | 147.7 KB | Display | PDB format |
| PDBx/mmJSON format | 2yvj.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/yv/2yvjftp://data.pdbj.org/pub/pdb/validation_reports/yv/2yvj | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2e4pC  2e4qC  2gqwC  2gr0C  2yvfC  2yvgC C: citing same article ( |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj





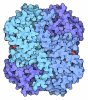




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 2 types, 3 molecules APB
| #1: Protein | Mass: 43221.184 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Pseudomonas sp. (bacteria) / Strain: strain KKS102 / Gene: bphA4 / Production host: #2: Protein | | Mass: 11926.499 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Pseudomonas sp. (bacteria) / Strain: strain KKS102 / Gene: bphA3 / Production host: |
|---|
-Non-polymers , 4 types, 83 molecules 
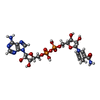
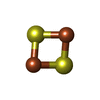

| #3: Chemical |  Mass: 785.550 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C27H33N9O15P2 / Comment: FAD*YM Mass: 785.550 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C27H33N9O15P2 / Comment: FAD*YM#4: Chemical |  Mass: 665.441 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H29N7O14P2 Mass: 665.441 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H29N7O14P2#5: Chemical | ChemComp-FES / |  Mass: 175.820 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe2S2 Mass: 175.820 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe2S2#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 78 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.94 Å3/Da / Density % sol: 58.1 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 6.5 Details: 0.1M MES, 30%(W/V) PEG4000, pH 6.50, VAPOR DIFFUSION, SITTING DROP, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Photon Factory  / Beamline: AR-NW12A / Wavelength: 0.978 / Beamline: AR-NW12A / Wavelength: 0.978 |
| Detector | Type: ADSC QUANTUM 210 / Detector: CCD / Date: Nov 12, 2005 |
| Radiation | Monochromator: SI(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.978 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→18 Å / Num. obs: 86196 / % possible obs: 96.7 % / Observed criterion σ(I): 0 / Redundancy: 9.3 % / Rmerge(I) obs: 0.082 / Net I/σ(I): 21.18 |
| Reflection shell | Resolution: 1.9→2 Å / Redundancy: 6.8 % / Rmerge(I) obs: 0.484 / Mean I/σ(I) obs: 5.46 / % possible all: 93.3 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.9→10 Å / Num. parameters: 28196 / Num. restraintsaints: 29665 / Cross valid method: FREE R / σ(F): 0 / Stereochemistry target values: Engh & Huber Details: THE CRYSTAL FORM IS TWINNED BY THE OPERATOR L,-K,H, PERFECT TWINED CRYSTAL.
| |||||||||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 0 / Occupancy sum hydrogen: 0 / Occupancy sum non hydrogen: 7044.4 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→10 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
|
