 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2yd1: Crystal structure of the N-terminal Ig1-2 module of Drosophila Re... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2yd1 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the N-terminal Ig1-2 module of Drosophila Receptor Protein Tyrosine Phosphatase DLAR | ||||||
 Components Components | TYROSINE-PROTEIN PHOSPHATASE LAR | ||||||
 Keywords Keywords | HYDROLASE | ||||||
| Function / homology |  Function and homology information Function and homology informationcentripetally migrating follicle cell migration / negative regulation of homophilic cell adhesion / Receptor-type tyrosine-protein phosphatases / positive regulation of plasma membrane bounded cell projection assembly / R7 cell development / Other semaphorin interactions / Synaptic adhesion-like molecules / photoreceptor cell morphogenesis / Insulin receptor recycling / NCAM signaling for neurite out-growth ...centripetally migrating follicle cell migration / negative regulation of homophilic cell adhesion / Receptor-type tyrosine-protein phosphatases / positive regulation of plasma membrane bounded cell projection assembly / R7 cell development / Other semaphorin interactions / Synaptic adhesion-like molecules / photoreceptor cell morphogenesis / Insulin receptor recycling / NCAM signaling for neurite out-growth / RAF/MAP kinase cascade / regulation of axon extension involved in axon guidance / SAM domain binding / axon target recognition / synaptic assembly at neuromuscular junction / hematopoietic stem cell homeostasis / transmembrane receptor protein tyrosine phosphatase activity / Neutrophil degranulation / axon extension / motor neuron axon guidance / retinal ganglion cell axon guidance / positive regulation of filopodium assembly / oogenesis / cell leading edge / protein-tyrosine-phosphatase / negative regulation of insulin receptor signaling pathway / protein tyrosine phosphatase activity / basal plasma membrane / axon guidance / insulin receptor binding / nervous system development / heparin binding / spermatogenesis / cell adhesion / axon / focal adhesion / cell surface / signal transduction Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.8 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.8 Å | ||||||
 Authors Authors | Coles, C.H. / Shen, Y. / Tenney, A.P. / Siebold, C. / Sutton, G.C. / Lu, W. / Gallagher, J.T. / Jones, E.Y. / Flanagan, J.G. / Aricescu, A.R. | ||||||
 Citation Citation | Journal: Science / Year: 2011 Title: Proteoglycan-Specific Molecular Switch for Rptp Sigma Clustering and Neuronal Extension. Authors: Coles, C.H. / Shen, Y. / Tenney, A.P. / Siebold, C. / Sutton, G.C. / Lu, W. / Gallagher, J.T. / Jones, E.Y. / Flanagan, J.G. / Aricescu, A.R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2yd1.cif.gz | 91.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2yd1.ent.gz | 68.3 KB | Display | PDB format |
| PDBx/mmJSON format | 2yd1.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/yd/2yd1ftp://data.pdbj.org/pub/pdb/validation_reports/yd/2yd1 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2yd2C  2yd3C  2yd4SC  2yd5C  2yd6C  2yd7C  2yd8C  2yd9C C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj











- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 23247.146 Da / Num. of mol.: 1 / Fragment: IG1-2, RESIDUES 33-232 Source method: isolated from a genetically manipulated source Source: (gene. exp.)  HOMO SAPIENS (human) / References: UniProt: P16621, protein-tyrosine-phosphatase HOMO SAPIENS (human) / References: UniProt: P16621, protein-tyrosine-phosphatase |
|---|---|
| #2: Chemical | ChemComp-GLY / 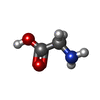  Type: peptide linking / Mass: 75.067 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H5NO2 Type: peptide linking / Mass: 75.067 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H5NO2 |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 116 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 116 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
| Sequence details | THE N-TERMINAL THREE AMINO ACID RESIDUES (ETG) AND THE C- TERMINAL NINE AMINO ACID RESIDUES ...THE N-TERMINAL THREE AMINO ACID RESIDUES (ETG) AND THE C- TERMINAL NINE AMINO ACID RESIDUES (GTKHHHHHH) ARE DERIVED FROM THE PHLSEC VECTOR |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.83 Å3/Da / Density % sol: 34 % / Description: NONE |
|---|---|
| Crystal grow | pH: 5 Details: 25% W/V PEG 1500, 0.1M SPG SYSTEM, 35MM AMMONIUM SULPHATE, PH 5.0 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID23-2 / Wavelength: 0.8726 / Beamline: ID23-2 / Wavelength: 0.8726 |
| Detector | Type: MARMOSAIC 225 mm CCD / Detector: CCD |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.8726 Å / Relative weight: 1 |
| Reflection | Resolution: 1.8→50 Å / Num. obs: 16271 / % possible obs: 100 % / Observed criterion σ(I): 0 / Redundancy: 5.6 % / Biso Wilson estimate: 20.29 Å2 / Rmerge(I) obs: 0.08 / Net I/σ(I): 19.6 |
| Reflection shell | Resolution: 1.8→1.85 Å / Redundancy: 5.4 % / Rmerge(I) obs: 0.53 / Mean I/σ(I) obs: 3.1 / % possible all: 100 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2YD4 Resolution: 1.8→47.175 Å / SU ML: 0.21 / σ(F): 0 / Phase error: 25.46 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 40 Å2 / ksol: 0.376 e/Å3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 31.96 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.8→47.175 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
