 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2xs3 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of karilysin catalytic MMP domain | ||||||
 Components Components |
| ||||||
 Keywords Keywords | HYDROLASE / BACTERIAL MMP / VIRULENCE FACTOR / METALLOPROTEASE / ZINC-DEPENDENT / PEPTIDASE | ||||||
| Function / homology |  Function and homology information Function and homology informationHydrolases; Acting on peptide bonds (peptidases); Metalloendopeptidases / collagen catabolic process / extracellular matrix organization / metalloendopeptidase activity / extracellular matrix / proteolysis / extracellular region / zinc ion binding Similarity search - Function | ||||||
| Biological species |  TANNERELLA FORSYTHIA (bacteria) TANNERELLA FORSYTHIA (bacteria)SYNTHETIC CONSTRUCT (others) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.4 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.4 Å | ||||||
 Authors Authors | Cerda-Costa, N. / Guevara, T. / Karim, A.Y. / Ksiazek, M. / Nguyen, K.-A. / Arolas, J.L. / Potempa, J. / Gomis-Ruth, F.X. | ||||||
 Citation Citation | Journal: Mol.Microbiol. / Year: 2011 Title: The Structure of the Catalytic Domain of Tannerella Forsythia Karilysin Reveals It is a Bacterial Xenologue of Animal Matrix Metalloproteinases. Authors: Cerda-Costa, N. / Guevara, T. / Karim, A.Y. / Ksiazek, M. / Nguyen, K.A. / Arolas, J.L. / Potempa, J. / Gomis-Ruth, F.X. #1: Journal: Biol.Chem. / Year: 2010 Title: A Novel Matrix Metalloprotease-Like Enzyme (Karilysin) of the Periodontal Pathogen Tannerella Forsythia Atcc 43037. Authors: Karim, A.Y. / Kulczycka, M. / Kantyka, T. / Dubin, G. / Jabaiah, A. / Daugherty, P.S. / Thogersen, I.B. / Enghild, J.J. / Nguyen, K. / Potempa, J. #2: Journal: J.Innate.Immun. / Year: 2010 Title: Proteolytic Inactivation of Ll-37 by Karilysin, a Novel Virulence Mechanism of Tannerella Forsythia. Authors: Koziel, J. / Karim, A.Y. / Przybyszewska, K. / Ksiazek, M. / Rapala-Kozik, M. / Nguyen, K. / Potempa, J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2xs3.cif.gz | 155.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2xs3.ent.gz | 122.5 KB | Display | PDB format |
| PDBx/mmJSON format | 2xs3.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/xs/2xs3ftp://data.pdbj.org/pub/pdb/validation_reports/xs/2xs3 | HTTPS FTP |
|---|
-Related structure data
| Related structure data | 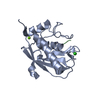 2xs4C  1mncS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 18703.615 Da / Num. of mol.: 2 / Fragment: RESIDUES 35-200 Source method: isolated from a genetically manipulated source Source: (gene. exp.) TANNERELLA FORSYTHIA (bacteria) / Production host: #2: Protein/peptide | Mass: 424.448 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Source: (natural) SYNTHETIC CONSTRUCT (others) #3: Chemical | ChemComp-ZN /   Mass: 65.409 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Zn#4: Chemical | ChemComp-TRS / |   Mass: 122.143 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H12NO3 / Comment: pH buffer*YM Mass: 122.143 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H12NO3 / Comment: pH buffer*YM#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 115 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 115 / Source method: isolated from a natural source / Formula: H2ONonpolymer details | ZINC CATION (ZN): CATALYTIC 999 AND STRUCTURAL | Sequence details | ONLY THE CATALYTIC DOMAIN (TYR35-PHE200) WAS CRYSTALLIZED. A PEPTIDE WAS FOUND AT THE ACTIVE SITE ...ONLY THE CATALYTIC DOMAIN (TYR35-PHE200) WAS CRYSTALLIZ | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.54 Å3/Da / Density % sol: 51.51 % / Description: NONE |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop Details: CRYSTALLIZATION ASSAYS WERE PERFORMED BY THE SITTING-DROP VAPOR DIFFUSION METHOD. RESERVOIR SOLUTIONS WERE PREPARED BY A TECAN ROBOT AND CRYSTALLIZATION DROPS WERE DISPENSED ON 96X2-WELL MRC ...Details: CRYSTALLIZATION ASSAYS WERE PERFORMED BY THE SITTING-DROP VAPOR DIFFUSION METHOD. RESERVOIR SOLUTIONS WERE PREPARED BY A TECAN ROBOT AND CRYSTALLIZATION DROPS WERE DISPENSED ON 96X2-WELL MRC PLATES (INNOVADYNE) BY A CARTESIAN (GENOMIC SOLUTIONS) NANODROP ROBOT AT THE HIGH-THROUGHPUT CRYSTALLOGRAPHY PLATFORM AT BARCELONA SCIENCE PARK. CRYSTALS SUITABLE FOR STRUCTURE ANALYSIS WERE OBTAINED FOR UNBOUND KLY18 IN A BRUKER STEADY-TEMPERATURE CRYSTAL FARM AT 20C FROM DROPS CONTAINING 100NL OF PROTEIN SOLUTION (AT 9MG ML-1 IN 5MM TRIS-HCL, PH8.0, 0.02% SODIUM AZIDE) AND 100NL OF 45% 2-METHYL-2,4-PENTANEDIOL, 0.2M AMMONIUM ACETATE, 0.1M TRIS-HCL, PH8.5 AS RESERVOIR SOLUTION. |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID23-2 / Wavelength: 0.8726 / Beamline: ID23-2 / Wavelength: 0.8726 |
| Detector | Type: MARRESEARCH / Detector: CCD |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.8726 Å / Relative weight: 1 |
| Reflection | Resolution: 2.4→45.3 Å / Num. obs: 15411 / % possible obs: 99.3 % / Observed criterion σ(I): 0 / Redundancy: 4.1 % / Biso Wilson estimate: 38.4 Å2 / Rmerge(I) obs: 0.09 / Net I/σ(I): 14.8 |
| Reflection shell | Resolution: 2.4→2.53 Å / Redundancy: 3.4 % / Rmerge(I) obs: 0.51 / Mean I/σ(I) obs: 2.5 / % possible all: 95.9 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1MNC Resolution: 2.4→50 Å / Cor.coef. Fo:Fc: 0.943 / Cor.coef. Fo:Fc free: 0.897 / SU B: 17.868 / SU ML: 0.203 / Cross valid method: THROUGHOUT / ESU R: 0.422 / ESU R Free: 0.277 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 33.731 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.4→50 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|