Entry Database : PDB / ID : 2xklTitle Crystal Structure of Mouse Apolipoprotein M APOLIPOPROTEIN M Keywords Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / Biological species MUS MUSCULUS (house mouse)Method / / / Resolution : 2.5 Å Authors Sevvana, M. / Kassler, K. / Josefin, A. / Weiler, S. / Dahlback, B. / Sticht, H. / Muller, Y.A. Journal : J.Mol.Biol. / Year : 2010Title : Mouse Apom Displays an Unprecedented Seven-Stranded Lipocalin Fold: Folding Decoy or Alternative Native Fold?Authors : Sevvana, M. / Kassler, K. / Ahnstrom, J. / Weiler, S. / Dahlback, B. / Sticht, H. / Muller, Y.A. History Deposition Jul 9, 2010 Deposition site / Processing site Revision 1.0 Oct 13, 2010 Provider / Type Revision 1.1 May 8, 2011 Group Revision 1.2 Jul 13, 2011 Group Revision 1.3 Dec 20, 2023 Group Data collection / Database references ... Data collection / Database references / Derived calculations / Other / Refinement description Category chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_database_status / pdbx_initial_refinement_model / struct_conn / struct_sheet / struct_site Item _database_2.pdbx_DOI / _database_2.pdbx_database_accession ... _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_database_status.status_code_sf / _struct_conn.ptnr1_auth_comp_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_asym_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr1_label_comp_id / _struct_conn.ptnr1_label_seq_id / _struct_conn.ptnr2_auth_comp_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_conn.ptnr2_label_atom_id / _struct_conn.ptnr2_label_comp_id / _struct_conn.ptnr2_label_seq_id / _struct_sheet.number_strands / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id Revision 1.4 Nov 13, 2024 Group / Category / pdbx_modification_feature
Show all Show less
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads








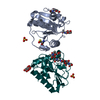

 PDBj
PDBj

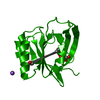
 Assembly
Assembly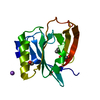





 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3
Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 60.095 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O
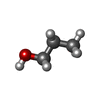
Mass: 60.095 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2
Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na
Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na Sample preparation
Sample preparation / Type:
/ Type:  Processing
Processing