 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2xem: Induced-fit and allosteric effects upon polyene binding revealed ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2xem | ||||||
|---|---|---|---|---|---|---|---|
| Title | Induced-fit and allosteric effects upon polyene binding revealed by crystal structures of the Dynemicin thioesterase | ||||||
 Components Components | DYNE7 | ||||||
 Keywords Keywords | BIOSYNTHETIC PROTEIN / POLYKETIDE BIOSYNTHESIS / ENEDIYNE ANTITUMOR AGENT / THIOESTERASE | ||||||
| Function / homology | Thioesterase-like superfamily / Hotdog Thioesterase / Thiol Ester Dehydrase; Chain A / HotDog domain superfamily / Roll / Alpha Beta / Chem-SSV / DynE7 Function and homology information Function and homology information | ||||||
| Biological species |  MICROMONOSPORA CHERSINA (bacteria) MICROMONOSPORA CHERSINA (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.1 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.1 Å | ||||||
 Authors Authors | Liew, C.W. / Sharff, A. / Kotaka, M. / Kong, R. / Bricogne, G. / Liang, Z.X. / Lescar, J. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2010 Title: Induced-Fit Upon Ligand Binding Revealed by Crystal Structures of the Hot-Dog Fold Thioesterase in Dynemicin Biosynthesis. Authors: Liew, C.W. / Sharff, A. / Kotaka, M. / Kong, R. / Sun, H. / Qureshi, I. / Bricogne, G. / Liang, Z.X. / Lescar, J. #1: Journal: J.Biol.Chem. / Year: 2009Title: Structure and Catalytic Mechanism of the Thioesterase Cale7 in Enediyne Biosynthesis. Authors: Kotaka, M. / Kong, R. / Qureshi, I. / Ho, Q.S. / Sun, H. / Liew, C.W. / Goh, L.P. / Cheung, P. / Mu, Y. / Lescar, J. / Liang, Z. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2xem.cif.gz | 243.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2xem.ent.gz | 196.4 KB | Display | PDB format |
| PDBx/mmJSON format | 2xem.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/xe/2xemftp://data.pdbj.org/pub/pdb/validation_reports/xe/2xem | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2xflC  2w3xS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 16937.232 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.) MICROMONOSPORA CHERSINA (bacteria) / Plasmid: PCDF-2 LIC/EK / Production host: #2: Chemical | ChemComp-SSV / ( | 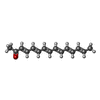  Mass: 214.303 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H18O Mass: 214.303 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H18O#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 546 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 546 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.61 Å3/Da / Density % sol: 53 % / Description: NONE |
|---|---|
| Crystal grow | pH: 7.5 / Details: PH 7.5 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSRRC  / Beamline: BL13B1 / Wavelength: 1.072 / Beamline: BL13B1 / Wavelength: 1.072 |
| Detector | Type: ADSC CCD / Detector: CCD / Details: MIRROR |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.072 Å / Relative weight: 1 |
| Reflection | Resolution: 2.1→15 Å / Num. obs: 39917 / % possible obs: 97.3 % / Observed criterion σ(I): 2.4 / Redundancy: 3.7 % / Biso Wilson estimate: 30.41 Å2 / Rmerge(I) obs: 0.08 / Net I/σ(I): 7 |
| Reflection shell | Resolution: 2.1→6.6 Å / Redundancy: 3.8 % / Rmerge(I) obs: 0.5 / Mean I/σ(I) obs: 2.4 / % possible all: 97 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2W3X Resolution: 2.1→14.84 Å / Cor.coef. Fo:Fc: 0.9547 / Cor.coef. Fo:Fc free: 0.9432 / SU R Cruickshank DPI: 0.172 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.184 / SU Rfree Blow DPI: 0.149 / SU Rfree Cruickshank DPI: 0.146 / Details: IDEAL-DISTANCE CONTACT TERM SETUP.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 34.6 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.205 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.1→14.84 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.1→2.15 Å / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
