 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2w75: Structures of P. aeruginosa FpvA bound to heterologous pyoverdine... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2w75 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structures of P. aeruginosa FpvA bound to heterologous pyoverdines: Apo-FpvA | ||||||
 Components Components | FERRIPYOVERDINE RECEPTOR | ||||||
 Keywords Keywords | RECEPTOR / TONB BOX / TRANSPORT / SIDEROPHORE / CELL MEMBRANE / ION TRANSPORT / IRON TRANSPORT / TONB-DEPENDENT TRANSPORTER | ||||||
| Function / homology |  Function and homology information Function and homology informationpyoverdine biosynthetic process / siderophore-iron import into cell / siderophore uptake transmembrane transporter activity / siderophore transport / cell outer membrane / signaling receptor activity / membrane Similarity search - Function | ||||||
| Biological species |   PSEUDOMONAS AERUGINOSA (bacteria) PSEUDOMONAS AERUGINOSA (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.9 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.9 Å | ||||||
 Authors Authors | Greenwald, J. / Nader, M. / Celia, H. / Gruffaz, C. / Meyer, J.-M. / Schalk, I.J. / Pattus, F. | ||||||
 Citation Citation | Journal: Mol.Microbiol. / Year: 2009 Title: Fpva Bound to Non-Cognate Pyoverdines: Molecular Basis of Siderophore Recognition by an Iron Transporter. Authors: Greenwald, J. / Nader, M. / Celia, H. / Gruffaz, C. / Geoffroy, V. / Meyer, J.M. / Schalk, I.J. / Pattus, F. | ||||||
| History |
| ||||||
| Remark 700 | SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "BE" IN EACH CHAIN ON SHEET RECORDS BELOW ... SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "BE" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 22-STRANDED BARREL THIS IS REPRESENTED BY A 23-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN THE SHEET RECORDS BELOW, TWO SHEETS ARE DEFINED. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2w75.cif.gz | 305.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2w75.ent.gz | 246.2 KB | Display | PDB format |
| PDBx/mmJSON format | 2w75.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/w7/2w75ftp://data.pdbj.org/pub/pdb/validation_reports/w7/2w75 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2w16C  2w6tC  2w6uC  2w76C  2w77C  2w78C  2o5pS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj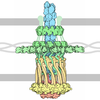
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 86556.086 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) PSEUDOMONAS AERUGINOSA (bacteria) / Strain: PAO1 / Plasmid: PPVR2 / Production host: PSEUDOMONAS AERUGINOSA (bacteria) / Strain (production host): K691 / References: UniProt: P48632#2: Chemical | 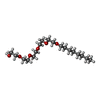  Mass: 350.491 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C18H38O6 / Comment: C8E5, detergent*YM Mass: 350.491 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C18H38O6 / Comment: C8E5, detergent*YM#3: Chemical | ChemComp-PO4 /   Mass: 94.971 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: PO4 Mass: 94.971 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: PO4Sequence details | SIGNAL PEPTIDE (RESIDUES 1-43) IS NOT IN THE CRYSTALLIZ | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.92 Å3/Da / Density % sol: 68.41 % / Description: NONE |
|---|---|
| Crystal grow | Details: 1.3M NAH2PO4, 0.1M MES, PH 6.5 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: BW7B / Wavelength: 0.8423 / Beamline: BW7B / Wavelength: 0.8423 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.8423 Å / Relative weight: 1 |
| Reflection | Resolution: 2.9→107 Å / Num. obs: 53997 / % possible obs: 92.3 % / Redundancy: 2 % / Rmerge(I) obs: 0.1 / Net I/σ(I): 1.9 |
| Reflection shell | Resolution: 2.9→3.06 Å / Redundancy: 2 % / Rmerge(I) obs: 0.26 / Mean I/σ(I) obs: 1.9 / % possible all: 93.3 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2O5P Resolution: 2.9→107.21 Å / Cor.coef. Fo:Fc: 0.892 / Cor.coef. Fo:Fc free: 0.859 / SU B: 29.438 / SU ML: 0.26 / TLS residual ADP flag: UNVERIFIED / Cross valid method: THROUGHOUT / ESU R: 1.475 / ESU R Free: 0.376 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 9.32 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.9→107.21 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|