 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2w3o: Crystal structure of the human PNKP FHA domain in complex with an... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2w3o | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the human PNKP FHA domain in complex with an XRCC1-derived phosphopeptide | ||||||
 Components Components |
| ||||||
 Keywords Keywords | HYDROLASE / TRANSFERASE/PEPTIDE / FHA / PNKP / XRCC1 / KINASE / NUCLEUS / POLYNUCLEOTIDE KINASE 3' PHOSPHATASE / DNA DAMAGE / DNA REPAIR / TRANSFERASE / ATP-BINDING / MULTIFUNCTIONAL ENZYME / POLYMORPHISM / PHOSPHOPROTEIN / PHOSPHO- PEPTIDE / NUCLEOTIDE-BINDING / BASE EXCISION REPAIR / TRANSFERASE-PEPTIDE complex | ||||||
| Function / homology |  Function and homology information Function and homology informationpurine nucleotide binding / polynucleotide 3'-phosphatase / oxidized DNA binding / polynucleotide 3'-phosphatase activity / polynucleotide 5'-hydroxyl-kinase / ATP-dependent polydeoxyribonucleotide 5'-hydroxyl-kinase activity / telomeric DNA-containing double minutes formation / ERCC4-ERCC1 complex / negative regulation of protection from non-homologous end joining at telomere / ADP-D-ribose modification-dependent protein binding ...purine nucleotide binding / polynucleotide 3'-phosphatase / oxidized DNA binding / polynucleotide 3'-phosphatase activity / polynucleotide 5'-hydroxyl-kinase / ATP-dependent polydeoxyribonucleotide 5'-hydroxyl-kinase activity / telomeric DNA-containing double minutes formation / ERCC4-ERCC1 complex / negative regulation of protection from non-homologous end joining at telomere / ADP-D-ribose modification-dependent protein binding / negative regulation of protein ADP-ribosylation / poly-ADP-D-ribose binding / regulation of base-excision repair / single strand break repair / HDR through MMEJ (alt-NHEJ) / 3' overhang single-stranded DNA endonuclease activity / response to hydroperoxide / Resolution of AP sites via the single-nucleotide replacement pathway / positive regulation of double-strand break repair via nonhomologous end joining / APEX1-Independent Resolution of AP Sites via the Single Nucleotide Replacement Pathway / positive regulation of telomere maintenance / regulation of DNA repair / site of DNA damage / base-excision repair, gap-filling / Gap-filling DNA repair synthesis and ligation in GG-NER / hippocampus development / nucleotide-excision repair / response to radiation / base-excision repair / DNA-templated DNA replication / double-strand break repair via nonhomologous end joining / Gap-filling DNA repair synthesis and ligation in TC-NER / double-strand break repair / site of double-strand break / response to oxidative stress / double-stranded DNA binding / endonuclease activity / damaged DNA binding / chromosome, telomeric region / DNA repair / chromatin / nucleolus / enzyme binding / nucleoplasm / ATP binding / membrane / nucleus Similarity search - Function | ||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.85 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.85 Å | ||||||
 Authors Authors | Oliver, A.W. / Ali, A.A.E. / Pearl, L.H. | ||||||
 Citation Citation | Journal: Nucleic Acids Res. / Year: 2009 Title: Specific Recognition of a Multiply Phosphorylated Motif in the DNA Repair Scaffold Xrcc1 by the Fha Domain of Human Pnk. Authors: Ali, A.A.E. / Jukes, R.M. / Pearl, L.H. / Oliver, A.W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2w3o.cif.gz | 64.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2w3o.ent.gz | 46.9 KB | Display | PDB format |
| PDBx/mmJSON format | 2w3o.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/w3/2w3oftp://data.pdbj.org/pub/pdb/validation_reports/w3/2w3o | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2brfSC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |












-Links
 PDBj
PDBj







- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 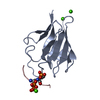
| ||||||||||||
| 2 | 
| ||||||||||||
| Unit cell |
| ||||||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 12117.620 Da / Num. of mol.: 2 / Fragment: FHA DOMAIN, RESIDUES 1-110 / Mutation: YES Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Description: SYNTHETIC GENE / Plasmid: PTWO-E / Production host:  References: UniProt: Q96T60, polynucleotide 3'-phosphatase, polynucleotide 5'-hydroxyl-kinase #2: Protein/peptide | Mass: 1015.764 Da / Num. of mol.: 2 / Fragment: RESIDUES 515-522 / Source method: obtained synthetically / Source: (synth.) HOMO SAPIENS (human) / References: UniProt: P18887#3: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Ca#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 286 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 286 / Source method: isolated from a natural source / Formula: H2OCompound details | ENGINEERED | Has protein modification | Y | Sequence details | THE SEQUENCE THAT WAS SYNTHESIZED WAS TAKEN FROM THAT REPORTED IN "MOLECULAR CLONING OF THE HUMAN ...THE SEQUENCE THAT WAS SYNTHESIZE | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.63 Å3/Da / Density % sol: 53.33 % / Description: NONE |
|---|---|
| Crystal grow | pH: 8.5 Details: 0.1 M TRIS-HCL PH 8.5, 0.25 M CACL2, 28% W/V PEG 4000, 0.2 M NDSB-221 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I04 / Wavelength: 1.208 / Beamline: I04 / Wavelength: 1.208 |
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Aug 4, 2008 / Details: KIRKPATRICK BAEZ BIMORPH MIRROR PAIR |
| Radiation | Monochromator: SI(111) DOUBLE CRYSTAL MONOCHROMATOR / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.208 Å / Relative weight: 1 |
| Reflection | Resolution: 1.85→38.75 Å / Num. obs: 19211 / % possible obs: 99 % / Observed criterion σ(I): 0 / Redundancy: 3.37 % / Rmerge(I) obs: 0.07 / Net I/σ(I): 5.78 |
| Reflection shell | Resolution: 1.85→1.95 Å / Redundancy: 2.12 % / Rmerge(I) obs: 0.3 / Mean I/σ(I) obs: 2.43 / % possible all: 95.3 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2BRF Resolution: 1.85→62.75 Å / Cor.coef. Fo:Fc: 0.963 / Cor.coef. Fo:Fc free: 0.941 / SU B: 3.452 / SU ML: 0.103 / Cross valid method: THROUGHOUT / ESU R: 0.145 / ESU R Free: 0.146 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 27.33 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.85→62.75 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|