| Entry | Database: PDB / ID: 2v59
|
|---|
| Title | CRYSTAL STRUCTURE OF BIOTIN CARBOXYLASE FROM E.COLI IN COMPLEX WITH POTENT INHIBITOR 2 |
|---|
 Components Components | BIOTIN CARBOXYLASE |
|---|
 Keywords Keywords | LIGASE / FATTY ACID BIOSYNTHESIS / BIOTIN CARBOXYLASE / NUCLEOTIDE-BINDING / ATP-BINDING / ANTIBACTERIAL / LIPID SYNTHESIS / FAS / BIOTIN / BACTERIAL / INHIBITOR |
|---|
| Function / homology |  Function and homology information Function and homology information
biotin carboxylase / malonyl-CoA biosynthetic process / biotin carboxylase activity / acetyl-CoA carboxylase activity / negative regulation of fatty acid biosynthetic process / fatty acid biosynthetic process / protein homodimerization activity / ATP binding / metal ion binding / cytosol / cytoplasmSimilarity search - Function Acetyl-CoA carboxylase, biotin carboxylase / : / Rossmann fold - #20 / Biotin carboxylase-like, N-terminal domain / Biotin carboxylase, C-terminal / Biotin carboxylation domain / Biotin carboxylase, N-terminal domain / Biotin carboxylase C-terminal domain / Biotin carboxylation domain profile. / Biotin carboxylase C-terminal domain ...Acetyl-CoA carboxylase, biotin carboxylase / : / Rossmann fold - #20 / Biotin carboxylase-like, N-terminal domain / Biotin carboxylase, C-terminal / Biotin carboxylation domain / Biotin carboxylase, N-terminal domain / Biotin carboxylase C-terminal domain / Biotin carboxylation domain profile. / Biotin carboxylase C-terminal domain / ATP-grasp fold, A domain / Carbamoyl-phosphate synthase subdomain signature 1. / Carbamoyl-phosphate synthetase large subunit-like, ATP-binding domain / Carbamoyl-phosphate synthase L chain, ATP binding domain / Rudiment single hybrid motif / ATP-grasp fold, B domain / ATP-grasp fold, subdomain 1 / Pre-ATP-grasp domain superfamily / ATP-grasp fold / ATP-grasp fold profile. / D-amino Acid Aminotransferase; Chain A, domain 1 / Dna Ligase; domain 1 / Carbamoyl-phosphate synthase subdomain signature 2. / Rossmann fold / 2-Layer Sandwich / 3-Layer(aba) Sandwich / Alpha BetaSimilarity search - Domain/homology |
|---|
| Biological species |   ESCHERICHIA COLI (E. coli) ESCHERICHIA COLI (E. coli) |
|---|
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.4 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.4 Å |
|---|
 Authors Authors | Mochalkin, I. / Miller, J.R. |
|---|
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 2009
Title: A Class of Selective Antibacterials Derived from a Protein Kinase Inhibitor Pharmacophore.
Authors: Miller, J.R. / Dunham, S. / Mochalkin, I. / Banotai, C. / Bowman, M. / Buist, S. / Dunkle, B. / Hanna, D. / Harwood, H.J. / Huband, M.D. / Karnovsky, A. / Kuhn, M. / Limberakis, C. / Liu, J. ...Authors: Miller, J.R. / Dunham, S. / Mochalkin, I. / Banotai, C. / Bowman, M. / Buist, S. / Dunkle, B. / Hanna, D. / Harwood, H.J. / Huband, M.D. / Karnovsky, A. / Kuhn, M. / Limberakis, C. / Liu, J.Y. / Mehrens, S. / Mueller, W.T. / Narasimhan, L. / Ogden, A. / Ohren, J. / Prasad, J.V. / Shelly, J.A. / Skerlos, L. / Sulavik, M. / Thomas, V.H. / Vanderroest, S. / Wang, L. / Wang, Z. / Whitton, A. / Zhu, T. / Stover, C.K. |
|---|
| History | | Deposition | Oct 2, 2008 | Deposition site: PDBE / Processing site: PDBE |
|---|
| Revision 1.0 | Jan 13, 2009 | Provider: repository / Type: Initial release |
|---|
| Revision 1.1 | Jul 13, 2011 | Group: Advisory / Version format compliance |
|---|
| Revision 1.2 | Dec 13, 2023 | Group: Data collection / Database references ...Data collection / Database references / Other / Refinement description
Category: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / pdbx_database_status / pdbx_initial_refinement_model
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_database_status.status_code_sf |
|---|
|
|---|
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads



















 PDBj
PDBj




 Assembly
Assembly


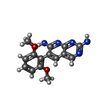
 Mass: 297.312 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C15H15N5O2
Mass: 297.312 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C15H15N5O2 Mass: 18.015 Da / Num. of mol.: 236 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 236 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: 17-BM / Wavelength: 1
/ Beamline: 17-BM / Wavelength: 1  Processing
Processing