 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2omb: Bence Jones KWR Protein- Immunoglobulin Light Chain Dimer, P3(1)2... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2omb | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Bence Jones KWR Protein- Immunoglobulin Light Chain Dimer, P3(1)21 Crystal Form | |||||||||
 Components Components | Bence Jones KWR Protein - Immunoglobulin Light Chain | |||||||||
 Keywords Keywords | IMMUNE SYSTEM / Immunoglobulin Light Chain | |||||||||
| Function / homology | Immunoglobulins / Immunoglobulin-like / Sandwich / Mainly Beta / PHENOL Function and homology information Function and homology information | |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.9 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.9 Å | |||||||||
 Authors Authors | Makino, D.L. / Larson, S.B. / McPherson, A. | |||||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2007 Title: Bence Jones KWR protein structures determined by X-ray crystallography. Authors: Makino, D.L. / Henschen-Edman, A.H. / Larson, S.B. / McPherson, A. | |||||||||
| History |
| |||||||||
| Remark 999 | SEQUENCE NO SUITABLE SEQUENCE DATABASE REFERENCE WAS AVAILABLE AT THE TIME OF PROCESSING THIS ENTRY |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2omb.cif.gz | 167.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2omb.ent.gz | 135.4 KB | Display | PDB format |
| PDBx/mmJSON format | 2omb.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/om/2ombftp://data.pdbj.org/pub/pdb/validation_reports/om/2omb | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2oldC  2omnC  1dclS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Antibody | Mass: 22698.914 Da / Num. of mol.: 4 / Source method: isolated from a natural source / Details: urine / Source: (natural) Homo sapiens (human) / Strain: KWR#2: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: SO4#3: Chemical | 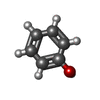  Mass: 94.111 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H6O Mass: 94.111 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H6O#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 16 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 16 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.52 Å3/Da / Density % sol: 65.03 % |
|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, sitting drop / pH: 8 Details: 1.5 M Ammonium Sulfate, 0.1M Tris/HCl pH8.0, VAPOR DIFFUSION, SITTING DROP, temperature 295K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL  / Beamline: BL7-1 / Wavelength: 1.08 Å / Beamline: BL7-1 / Wavelength: 1.08 Å |
| Detector | Type: MAR scanner 345 mm plate / Detector: IMAGE PLATE / Date: Jan 14, 2001 |
| Radiation | Monochromator: Si(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.08 Å / Relative weight: 1 |
| Reflection | Resolution: 2.9→100 Å / Num. all: 27674 / Num. obs: 27674 / % possible obs: 96.6 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 4.6 % / Biso Wilson estimate: 57.9 Å2 / Limit h max: 45 / Limit h min: 0 / Limit k max: 45 / Limit k min: 0 / Limit l max: 31 / Limit l min: 0 / Observed criterion F max: 699481.23 / Observed criterion F min: 0.45 / Rsym value: 0.1 / Net I/σ(I): 14.7 |
| Reflection shell | Resolution: 2.9→3.02 Å / Redundancy: 3.9 % / Mean I/σ(I) obs: 2.3 / Num. unique all: 2825 / Rsym value: 0.469 / % possible all: 90.1 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1DCL Resolution: 2.9→40 Å / Rfactor Rfree error: 0.007 / Occupancy max: 1 / Occupancy min: 1 / Isotropic thermal model: Isotropic / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / Stereochemistry target values: Engh & Huber Details: PARAMETERS FOR PYROGLUTAMIC ACID AND PHENOL WERE OBTAINED FROM HETERO-COMPOUND INFORMATION CENTRE - UPPSALA.
| |||||||||||||||||||||||||||
| Solvent computation | Solvent model: CNS bulk solvent model used / Bsol: 49.0414 Å2 / ksol: 0.347328 e/Å3 | |||||||||||||||||||||||||||
| Displacement parameters | Biso max: 147 Å2 / Biso mean: 68.3 Å2 / Biso min: 16.47 Å2
| |||||||||||||||||||||||||||
| Refine analyze |
| |||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.9→40 Å
| |||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.9→3 Å / Rfactor Rfree error: 0.029
| |||||||||||||||||||||||||||
| Xplor file |
|
