 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2nun | ||||||
|---|---|---|---|---|---|---|---|
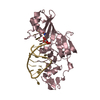
| Title | The structure of the type III effector AvrB complexed with ADP | ||||||
 Components Components | Avirulence B protein | ||||||
 Keywords Keywords | TOXIN/PROTEIN BINDING / type III effector / AvrB / nuclotide / pocket / ADP / TOXIN-PROTEIN BINDING COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  Pseudomonas syringae pv. glycinea (bacteria) Pseudomonas syringae pv. glycinea (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.4 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.4 Å | ||||||
 Authors Authors | Singer, A.U. / Desveaux, D. / Wu, A.J. / McNulty, B. / Dangl, J.L. / Sondek, J. | ||||||
 Citation Citation | Journal: Plos Pathog. / Year: 2007 Title: Type III Effector Activation via Nucleotide Binding, Phosphorylation, and Host Target Interaction. Authors: Desveaux, D. / Singer, A.U. / Wu, A.J. / McNulty, B.C. / Musselwhite, L. / Nimchuk, Z. / Sondek, J. / Dangl, J.L. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2nun.cif.gz | 77.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2nun.ent.gz | 56.3 KB | Display | PDB format |
| PDBx/mmJSON format | 2nun.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/nu/2nunftp://data.pdbj.org/pub/pdb/validation_reports/nu/2nun | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2nudC  1nh1S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | only one molecule in the asymmetric unit and the biological assembly |
-Components
| #1: Protein | Mass: 36181.262 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Pseudomonas syringae pv. glycinea (bacteria)Species: Pseudomonas savastanoi / Strain: pv glycinea / Gene: avrB / Plasmid: pProEX-HTa / Production host: |
|---|---|
| #2: Chemical | ChemComp-ADP /   Mass: 427.201 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM Mass: 427.201 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM |
| #3: Chemical | ChemComp-TRS /   Mass: 122.143 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H12NO3 / Comment: pH buffer*YM Mass: 122.143 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H12NO3 / Comment: pH buffer*YM |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 86 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 86 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.85 Å3/Da / Density % sol: 68.04 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, sitting drop / pH: 9 Details: Protein (8-10 mg/ml) was mixed with well solution (100 mM Glycine, pH 9.0, 20-30% PEG 550 MME)at a 1:1 ratio. Microseeding was employed to obtain better crystals. Nucleotides were ...Details: Protein (8-10 mg/ml) was mixed with well solution (100 mM Glycine, pH 9.0, 20-30% PEG 550 MME)at a 1:1 ratio. Microseeding was employed to obtain better crystals. Nucleotides were incorporated by exchanging drop and resevoir solutions with 20 l 27% PEG 500 MME and 100 mM Tris 7.5 plus 5 mM MgCl2, followed by a final exchange of this solution plus 5 mM nucleotide. Nucleotides were soaked for ~ 1 day. , VAPOR DIFFUSION, SITTING DROP, temperature 277K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 22-ID / Wavelength: 1 Å / Beamline: 22-ID / Wavelength: 1 Å |
| Detector | Type: MAR CCD 165 mm / Detector: CCD / Date: Oct 30, 2005 / Details: mirrors |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.15→20 Å / Num. all: 29955 / Num. obs: 24091 / % possible obs: 80.4 % / Observed criterion σ(F): 1 / Observed criterion σ(I): 1 / Redundancy: 4.9 % / Biso Wilson estimate: 45.1 Å2 / Rmerge(I) obs: 0.093 / Net I/σ(I): 28 |
| Reflection shell | Resolution: 2.15→2.23 Å / Redundancy: 3.3 % / Rmerge(I) obs: 0.318 / Mean I/σ(I) obs: 2.63 / Num. unique all: 943 / % possible all: 32 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entry 1NH1 Resolution: 2.4→20 Å / Rfactor Rfree error: 0.008 / Data cutoff high absF: 1928271.38 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 1.5 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 32.5504 Å2 / ksol: 0.30493 e/Å3 | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 52.8 Å2
| ||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.4→20 Å
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.4→2.55 Å / Rfactor Rfree error: 0.024 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||
| Xplor file |
|
