 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2i42: Crystal structure of Yersinia protein tyrosine phosphatase comple... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2i42 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of Yersinia protein tyrosine phosphatase complexed with vanadate, a transition state analogue | ||||||
 Components Components | Tyrosine-protein phosphatase | ||||||
 Keywords Keywords | HYDROLASE / Yersinia PTPase / vanadate / transition state analogue | ||||||
| Function / homology |  Function and homology information Function and homology informationprotein-tyrosine-phosphatase / protein tyrosine phosphatase activity / extracellular region Similarity search - Function | ||||||
| Biological species |  Yersinia enterocolitica (bacteria) Yersinia enterocolitica (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / FOURIER SYNTHESIS / Resolution: 2.2 Å X-RAY DIFFRACTION / FOURIER SYNTHESIS / Resolution: 2.2 Å | ||||||
 Authors Authors | Vijayalakshmi, J. / Saper, M.A. | ||||||
 Citation Citation | Journal: To be Published Title: Crystal structure of Yersinia protein tyrosine phosphatase complexed with vanadate, a transition state analogue Authors: Vijayalakshmi, J. / Saper, M.A. #1: Journal: PROC.NATL.ACAD.SCI.USA / Year: 1995 Title: Visualization of intermediate and transition-state structures in protein tyrosine phosphatase catalysis. Authors: Denu, J.M. / Lohse, D.L. / Vijayalakshmi, J. / Saper, M.A. / Dixon, J.E. #2: Journal: PROTEIN SCI. / Year: 1995Title: A ligand-induced conformational change in the Yersinia protein tyrosine phosphatase. Authors: Schubert, H.L. / Fauman, E.B. / Stuckey, J.A. / Dixon, J.E. / Saper, M.A. #3: Journal: Proc.Natl.Acad.Sci.USA / Year: 1994 Title: Dissecting the catalytic mechanism of protein tyrosine phosphatases. Authors: Zhang, Z.-Y. / Wang, Y. / Dixon, J.E. | ||||||
| History |
| ||||||
| Remark 600 | HETEROGEN VANADATE IS IN A TRIGONAL BIPYRAMIDAL GEOMETRY AND THE VANADIUM ATOM IS COVALENTLY BONDED ...HETEROGEN VANADATE IS IN A TRIGONAL BIPYRAMIDAL GEOMETRY AND THE VANADIUM ATOM IS COVALENTLY BONDED TO SG ATOM OF CYS 403 WITH A BONDING DISTANCE OF 2.52 A. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2i42.cif.gz | 73.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2i42.ent.gz | 52.7 KB | Display | PDB format |
| PDBx/mmJSON format | 2i42.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/i4/2i42ftp://data.pdbj.org/pub/pdb/validation_reports/i4/2i42 | HTTPS FTP |
|---|
-Related structure data












-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 33553.883 Da / Num. of mol.: 1 / Fragment: Tyrosine-protein phosphatase / Mutation: C235R Source method: isolated from a genetically manipulated source Source: (gene. exp.) Yersinia enterocolitica (bacteria) / Strain: W22703 / Gene: yopH, yop51 / Plasmid: pT7-7 / Species (production host): Escherichia coli / Production host: |
|---|---|
| #2: Chemical | ChemComp-VO4 / 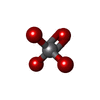  Mass: 114.939 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: VO4 Mass: 114.939 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: VO4 |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 183 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 183 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.26 Å3/Da / Density % sol: 41.25 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 8.5 Details: 400 mM NaCl, 14% PEG 4K, 5% 2-proponol .15% BME, 100mM Tris HCl pH 8.5, 1mM vanadate, VAPOR DIFFUSION, HANGING DROP, temperature 298K |
-Data collection
| Diffraction | Mean temperature: 298 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 Å |
| Detector | Type: SDMS / Detector: AREA DETECTOR / Date: Jan 1, 1995 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.1→10 Å / Num. all: 13593 / Num. obs: 13593 / % possible obs: 82 % / Observed criterion σ(I): 0 / Redundancy: 4.02 % / Rmerge(I) obs: 0.079 |
| Reflection shell | Resolution: 2.1→2.3 Å / Redundancy: 2.14 % / Rmerge(I) obs: 0.25 / Num. unique all: 1336 / % possible all: 74.3 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS Starting model: PDB entries 1YTW and 1YTS Resolution: 2.2→7 Å / Isotropic thermal model: Isotropic / σ(F): 2 / Stereochemistry target values: Engh & Huber Details: Residues Met163-Val186 were not included in the refinement due to missing electron densities.
| ||||||||||||||||||||||||
| Displacement parameters | Biso mean: 17.3 Å2 | ||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.23 Å / Luzzati d res low obs: 6 Å | ||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.2→7 Å
| ||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.2→2.3 Å
|
