 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2g25: E. Coli Pyruvate Dehydrogenase Phosphonolactylthiamin Diphosphate... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2g25 | ||||||
|---|---|---|---|---|---|---|---|
| Title | E. Coli Pyruvate Dehydrogenase Phosphonolactylthiamin Diphosphate Complex | ||||||
 Components Components | Pyruvate dehydrogenase E1 component | ||||||
 Keywords Keywords | OXIDOREDUCTASE / PLTHDP / PHOSPHONOLACTYLTHIAMIN DIPHOSPHATE / PYRUVATE DEHYDROGENASE E1 COMPONENT | ||||||
| Function / homology |  Function and homology information Function and homology informationpyruvate dehydrogenase (acetyl-transferring) / pyruvate dehydrogenase (acetyl-transferring) activity / pyruvate catabolic process / thiamine pyrophosphate binding / small molecule binding / molecular adaptor activity / magnesium ion binding / protein homodimerization activity / membrane / identical protein binding / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / DIFFERENCE FOURIER / Resolution: 2.1 Å X-RAY DIFFRACTION / DIFFERENCE FOURIER / Resolution: 2.1 Å | ||||||
 Authors Authors | Furey, W. / Arjunan, P. / Chandrasekhar, K. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2006 Title: A Thiamin-bound, Pre-decarboxylation Reaction Intermediate Analogue in the Pyruvate Dehydrogenase E1 Subunit Induces Large Scale Disorder-to-Order Transformations in the Enzyme and Reveals ...Title: A Thiamin-bound, Pre-decarboxylation Reaction Intermediate Analogue in the Pyruvate Dehydrogenase E1 Subunit Induces Large Scale Disorder-to-Order Transformations in the Enzyme and Reveals Novel Structural Features in the Covalently Bound Adduct. Authors: Arjunan, P. / Sax, M. / Brunskill, A. / Chandrasekhar, K. / Nemeria, N. / Zhang, S. / Jordan, F. / Furey, W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2g25.cif.gz | 347.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2g25.ent.gz | 276.2 KB | Display | PDB format |
| PDBx/mmJSON format | 2g25.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/g2/2g25ftp://data.pdbj.org/pub/pdb/validation_reports/g2/2g25 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2g28C  1l8aS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 99657.164 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) References: UniProt: P0AFG8, pyruvate dehydrogenase (acetyl-transferring) #2: Chemical |   Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg#3: Chemical | ChemComp-PO4 /   Mass: 94.971 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: PO4 Mass: 94.971 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: PO4#4: Chemical | 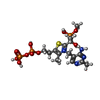  Mass: 563.373 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C15H26N4O11P3S Mass: 563.373 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C15H26N4O11P3S#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 564 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 564 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.34 Å3/Da / Density % sol: 47.38 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, sitting drop / pH: 7 Details: PEG2000 MONOMETHYL ETHER, PROPANOL, SODIUM AZIDE, HEPES BUFFER, MAGNESIUM CHLORIDE, PHOSPHONOLACTYL- THIAMIN DIPHOSPHATE, PH 7.00, VAPOR DIFFUSION, SITTING DROP, TEMPERATURE 291K |
-Data collection
| Diffraction | Mean temperature: 93 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 / Wavelength: 1.5418 Å |
| Detector | Type: SIEMENS HI-STAR / Detector: AREA DETECTOR / Date: Mar 25, 2002 / Details: Osmic Mirrors |
| Radiation | Monochromator: Osmic Mirror / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.1→40.5 Å / Num. all: 99699 / Num. obs: 99699 / % possible obs: 94 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 4.7 % / Rmerge(I) obs: 0.065 / Rsym value: 0.087 / Net I/σ(I): 24.82 |
| Reflection shell | Resolution: 2.1→2.19 Å / Mean I/σ(I) obs: 5.51 / % possible all: 60 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: DIFFERENCE FOURIER Starting model: PDB ENTRY 1L8A Resolution: 2.1→8 Å / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 2 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 10.55 Å2 | ||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.1→8 Å
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.1→2.19 Å / Total num. of bins used: 7
|
