RNA exonuclease activity / DNA ligase (ATP) / DNA ligase (ATP) activity / DNA biosynthetic process / nucleotidyltransferase activity / DNA recombination / DNA-directed DNA polymerase activity / DNA repair / DNA binding / ATP binding / metal ion binding 類似検索 - 分子機能
DNA ligase D / LigD polymerase domain, PaeLigD-type / DNA ligase D, ligase domain / DNA ligase D, polymerase domain / : / LigD, primase-polymerase domain / DNA ligase D, 3'-phosphoesterase domain / DNA polymerase Ligase (LigD) / DNA primase, PRIM domain / DNA primase, PRIM domain ...DNA ligase D / LigD polymerase domain, PaeLigD-type / DNA ligase D, ligase domain / DNA ligase D, polymerase domain / : / LigD, primase-polymerase domain / DNA ligase D, 3'-phosphoesterase domain / DNA polymerase Ligase (LigD) / DNA primase, PRIM domain / DNA primase, PRIM domain / DNA ligase, ATP-dependent, C-terminal / ATP dependent DNA ligase C terminal region / ATP-dependent DNA ligase family profile. / DNA ligase, ATP-dependent, central / ATP dependent DNA ligase domain / Nucleic acid-binding, OB-fold / Alpha-Beta Complex / Alpha Beta 類似検索 - ドメイン・相同性
2'-DEOXYADENOSINE 5'-TRIPHOSPHATE / : / Multifunctional non-homologous end joining protein LigD 類似検索 - 構成要素
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード 機能・相同性情報
機能・相同性情報
 Pseudomonas aeruginosa (緑膿菌)
Pseudomonas aeruginosa (緑膿菌) X線回折 /
X線回折 /  データ登録者
データ登録者 引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード











 PDBj
PDBj



 集合体
集合体



 分子量: 54.938 Da / 分子数: 4 / 由来タイプ: 合成 / 式: Mn
分子量: 54.938 Da / 分子数: 4 / 由来タイプ: 合成 / 式: Mn
 分子量: 96.063 Da / 分子数: 3 / 由来タイプ: 合成 / 式: SO4
分子量: 96.063 Da / 分子数: 3 / 由来タイプ: 合成 / 式: SO4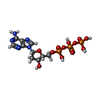
 分子量: 491.182 Da / 分子数: 2 / 由来タイプ: 合成 / 式: C10H16N5O12P3
分子量: 491.182 Da / 分子数: 2 / 由来タイプ: 合成 / 式: C10H16N5O12P3 分子量: 18.015 Da / 分子数: 495 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 495 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 / ビームライン: X29A / 波長: 0.9792 Å
/ ビームライン: X29A / 波長: 0.9792 Å 解析
解析