 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2ch6: Crystal structure of human N-acetylglucosamine kinase in complex ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2ch6 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of human N-acetylglucosamine kinase in complex with ADP and glucose | ||||||
 Components Components | N-ACETYL-D-GLUCOSAMINE KINASE | ||||||
 Keywords Keywords | TRANSFERASE / SUGAR KINASE / RIBONUCLEASE H FOLD / SUGAR KINASE/HSP70/ACTIN SUPERFAMILY | ||||||
| Function / homology |  Function and homology information Function and homology informationmuramyl dipeptide kinase activity / N-acetylmannosamine metabolic process / N-acetylglucosamine kinase / N-acylmannosamine kinase / N-acetylglucosamine kinase activity / N-acylmannosamine kinase activity / Synthesis of UDP-N-acetyl-glucosamine / N-acetylglucosamine catabolic process / N-acetylglucosamine metabolic process / Transferases; Transferring phosphorus-containing groups; Phosphotransferases with an alcohol group as acceptor ...muramyl dipeptide kinase activity / N-acetylmannosamine metabolic process / N-acetylglucosamine kinase / N-acylmannosamine kinase / N-acetylglucosamine kinase activity / N-acylmannosamine kinase activity / Synthesis of UDP-N-acetyl-glucosamine / N-acetylglucosamine catabolic process / N-acetylglucosamine metabolic process / Transferases; Transferring phosphorus-containing groups; Phosphotransferases with an alcohol group as acceptor / N-acetylneuraminate catabolic process / positive regulation of nucleotide-binding oligomerization domain containing 2 signaling pathway / UDP-N-acetylglucosamine biosynthetic process / response to muramyl dipeptide / defense response to bacterium / innate immune response / extracellular exosome / ATP binding / cytosol Similarity search - Function | ||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.72 Å X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.72 Å | ||||||
 Authors Authors | Weihofen, W.A. / Berger, M. / Chen, H. / Saenger, W. / Hinderlich, S. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2006 Title: Structures of Human N-Acetylglucosamine Kinase in Two Complexes with N-Acetylglucosamine and with Adp/Glucose: Insights Into Substrate Specificity and Regulation. Authors: Weihofen, W.A. / Berger, M. / Chen, H. / Saenger, W. / Hinderlich, S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2ch6.cif.gz | 256.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2ch6.ent.gz | 209.7 KB | Display | PDB format |
| PDBx/mmJSON format | 2ch6.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ch/2ch6ftp://data.pdbj.org/pub/pdb/validation_reports/ch/2ch6 | HTTPS FTP |
|---|
-Related structure data











-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||
| 2 | 
| ||||||||||||||||
| Unit cell |
| ||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper:
| ||||||||||||||||
| Details | THE ASYMMETRIC UNIT CONTAINS TWO NAGK DIMERS (CHAINS AD FORM ONE, AND CHAINS C AND B FORM THE OTHER). |
-Components
| #1: Protein | Mass: 37417.609 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human)Description: CDNA FROM THE RESOURCE CENTER OF THE GERMAN HUMAN GENOME PROJECT Plasmid: PGEX-2T / Production host:  #2: Chemical | ChemComp-ADP /   Mass: 427.201 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM Mass: 427.201 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM#3: Sugar | ChemComp-GLC / 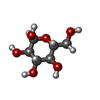  Type: D-saccharide, alpha linking / Mass: 180.156 Da / Num. of mol.: 4 Type: D-saccharide, alpha linking / Mass: 180.156 Da / Num. of mol.: 4Source method: isolated from a genetically manipulated source Formula: C6H12O6 |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.6 Å3/Da / Density % sol: 51.9 % Description: MAD DATA OF SEMET LABELED CRYSTALS WERE COLLECTED AT PROTEIN STRUCTURE FACTORY BEAMLINE BL2 OF FREE UNIVERSITY BERLIN AT BESSY |
|---|---|
| Crystal grow | pH: 5.6 Details: 100 MM MES PH5.6, 100 MM NACL, 2% (W/V) PEG 20K, pH 5.60 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-2 / Wavelength: 0.933 / Beamline: ID14-2 / Wavelength: 0.933 |
| Detector | Type: ADSC CCD / Detector: CCD / Date: Feb 1, 2005 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.933 Å / Relative weight: 1 |
| Reflection | Resolution: 2.72→30 Å / Num. obs: 40858 / % possible obs: 98.8 % / Observed criterion σ(I): 2 / Redundancy: 6 % / Rmerge(I) obs: 0.06 / Net I/σ(I): 20.2 |
| Reflection shell | Resolution: 2.72→2.96 Å / Redundancy: 4.2 % / Rmerge(I) obs: 0.38 / Mean I/σ(I) obs: 3.8 / % possible all: 98.6 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 2.72→29.8 Å / Cor.coef. Fo:Fc: 0.924 / Cor.coef. Fo:Fc free: 0.878 / SU B: 27.79 / SU ML: 0.32 / TLS residual ADP flag: UNVERIFIED / Cross valid method: THROUGHOUT / ESU R Free: 0.41 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 53.67 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.72→29.8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|