| Entry | Database: PDB / ID: 2cdz
|
|---|
| Title | CRYSTAL STRUCTURE OF THE HUMAN P21-ACTIVATED KINASE 4 IN COMPLEX WITH CGP74514A |
|---|
 Components Components | SERINE/THREONINE-PROTEIN KINASE PAK 4 |
|---|
 Keywords Keywords | TRANSFERASE / PROTEIN KINASE / STE20 / PAK4 / ATP-BINDING |
|---|
| Function / homology |  Function and homology information Function and homology information
dendritic spine development / cadherin binding involved in cell-cell adhesion / Activation of RAC1 / RHOV GTPase cycle / RHOJ GTPase cycle / RHOQ GTPase cycle / : / regulation of MAPK cascade / RHOU GTPase cycle / CDC42 GTPase cycle ...dendritic spine development / cadherin binding involved in cell-cell adhesion / Activation of RAC1 / RHOV GTPase cycle / RHOJ GTPase cycle / RHOQ GTPase cycle / : / regulation of MAPK cascade / RHOU GTPase cycle / CDC42 GTPase cycle / RHOH GTPase cycle / RHOG GTPase cycle / RAC2 GTPase cycle / RAC3 GTPase cycle / negative regulation of endothelial cell apoptotic process / cytoskeleton organization / RAC1 GTPase cycle / cellular response to starvation / adherens junction / regulation of cell growth / positive regulation of angiogenesis / cell migration / eukaryotic translation initiation factor 2alpha kinase activity / 3-phosphoinositide-dependent protein kinase activity / DNA-dependent protein kinase activity / ribosomal protein S6 kinase activity / histone H3S10 kinase activity / histone H2AXS139 kinase activity / histone H3S28 kinase activity / histone H4S1 kinase activity / histone H2BS14 kinase activity / histone H3T3 kinase activity / histone H2AS121 kinase activity / Rho-dependent protein serine/threonine kinase activity / histone H2BS36 kinase activity / histone H3S57 kinase activity / histone H2AT120 kinase activity / AMP-activated protein kinase activity / histone H2AS1 kinase activity / histone H3T6 kinase activity / histone H3T11 kinase activity / histone H3T45 kinase activity / non-specific serine/threonine protein kinase / protein kinase activity / intracellular signal transduction / protein serine kinase activity / focal adhesion / protein serine/threonine kinase activity / apoptotic process / Golgi apparatus / signal transduction / ATP binding / cytosol / cytoplasmSimilarity search - Function p21 activated kinase binding domain / : / CRIB domain superfamily / P21-Rho-binding domain / CRIB domain profile. / P21-Rho-binding domain / CRIB domain / Phosphorylase Kinase; domain 1 / Phosphorylase Kinase; domain 1 / Transferase(Phosphotransferase) domain 1 ...p21 activated kinase binding domain / : / CRIB domain superfamily / P21-Rho-binding domain / CRIB domain profile. / P21-Rho-binding domain / CRIB domain / Phosphorylase Kinase; domain 1 / Phosphorylase Kinase; domain 1 / Transferase(Phosphotransferase) domain 1 / Transferase(Phosphotransferase); domain 1 / Protein kinase domain / Protein kinase, ATP binding site / Protein kinases ATP-binding region signature. / Protein kinase domain profile. / Protein kinase domain / Protein kinase-like domain superfamily / 2-Layer Sandwich / Orthogonal Bundle / Mainly Alpha / Alpha BetaSimilarity search - Domain/homology |
|---|
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) |
|---|
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.3 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.3 Å |
|---|
 Authors Authors | Debreczeni, J.E. / Ugochukwu, E. / Eswaran, J. / Filippakopoulos, P. / Das, S. / Fedorov, O. / Sundstrom, M. / Arrowsmith, C. / Weigelt, J. / Edwards, A. ...Debreczeni, J.E. / Ugochukwu, E. / Eswaran, J. / Filippakopoulos, P. / Das, S. / Fedorov, O. / Sundstrom, M. / Arrowsmith, C. / Weigelt, J. / Edwards, A. / von Delft, F. / Knapp, S. |
|---|
 Citation Citation | Journal: Structure / Year: 2007
Title: Crystal Structures of the P21-Activated Kinases Pak4, Pak5, and Pak6 Reveal Catalytic Domain Plasticity of Active Group II Paks.
Authors: Eswaran, J. / Lee, W.H. / Debreczeni, J.E. / Filippakopoulos, P. / Turnbull, A. / Fedorov, O. / Deacon, S.W. / Peterson, J.R. / Knapp, S. |
|---|
| History | | Deposition | Jan 31, 2006 | Deposition site: PDBE / Processing site: PDBE |
|---|
| Revision 1.0 | Feb 8, 2006 | Provider: repository / Type: Initial release |
|---|
| Revision 1.1 | Jul 13, 2011 | Group: Advisory / Version format compliance |
|---|
| Revision 1.2 | Jan 24, 2018 | Group: Structure summary / Category: audit_author / Item: _audit_author.name |
|---|
| Revision 1.3 | Apr 4, 2018 | Group: Data collection / Category: diffrn_source / Item: _diffrn_source.type |
|---|
| Revision 1.4 | Dec 13, 2023 | Group: Data collection / Database references ...Data collection / Database references / Derived calculations / Other / Refinement description
Category: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / pdbx_database_status / pdbx_initial_refinement_model / struct_conn
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession ..._database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_database_status.status_code_sf / _struct_conn.pdbx_leaving_atom_flag |
|---|
| Revision 1.5 | Nov 6, 2024 | Group: Structure summary / Category: pdbx_entry_details / pdbx_modification_feature |
|---|
|
|---|
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads












 PDBj
PDBj





 Assembly
Assembly

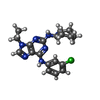
 Mass: 385.894 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H24ClN7
Mass: 385.894 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H24ClN7
 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4
 Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl
Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 18.015 Da / Num. of mol.: 117 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 117 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing