 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2cbr: CELLULAR RETINOIC ACID BINDING PROTEIN I IN COMPLEX WITH A RETINO... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2cbr | ||||||
|---|---|---|---|---|---|---|---|
| Title | CELLULAR RETINOIC ACID BINDING PROTEIN I IN COMPLEX WITH A RETINOBENZOIC ACID (AM80) | ||||||
 Components Components | PROTEIN (CRABP-I) | ||||||
 Keywords Keywords | TRANSPORT PROTEIN / RETINOIC-ACID TRANSPORT | ||||||
| Function / homology |  Function and homology information Function and homology informationRA biosynthesis pathway / retinoic acid binding / retinal binding / retinol binding / fatty acid transport / fatty acid binding / protein-containing complex / identical protein binding / nucleus / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.8 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.8 Å | ||||||
 Authors Authors | Chaudhuri, B. / Kleywegt, G.J. / Bergfors, T. / Jones, T.A. | ||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 1999 Title: Structures of cellular retinoic acid binding proteins I and II in complex with synthetic retinoids. Authors: Chaudhuri, B.N. / Kleywegt, G.J. / Broutin-L'Hermite, I. / Bergfors, T. / Senn, H. / Le Motte, P. / Partouche, O. / Jones, T.A. #1: Journal: Structure / Year: 1994Title: Crystal structures of cellular retinoic acid binding proteins I and II in complex with all-trans-retinoic acid and a synthetic retinoid. Authors: Kleywegt, G.J. / Bergfors, T. / Senn, H. / Le Motte, P. / Gsell, B. / Shudo, K. / Jones, T.A. #2: Journal: Adv.Protein Chem. / Year: 1994 Title: Lipid-binding proteins: a family of fatty acid and retinoid transport proteins. Authors: Banaszak, L. / Winter, N. / Xu, Z. / Bernlohr, D.A. / Cowan, S. / Jones, T.A. #3: Journal: Acta Crystallogr.,Sect.D / Year: 1994 Title: Crystallization and preliminary X-ray analysis of recombinant bovine cellular retinoic acid-binding protein. Authors: Bergfors, T. / Kleywegt, G.J. / Jones, T.A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2cbr.cif.gz | 39.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2cbr.ent.gz | 27 KB | Display | PDB format |
| PDBx/mmJSON format | 2cbr.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/cb/2cbrftp://data.pdbj.org/pub/pdb/validation_reports/cb/2cbr | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2cbsC  3cbsC  1cbrS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: generate Matrix: (0.231781, -0.961217, 0.149465), Vector: |
-Components
| #1: Protein | Mass: 15480.392 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Description: MUS MUSCULUS (IDENTICAL WITH BOVINE PROTEIN) RECOMBINANT GENE Gene: MOUSE / Plasmid: PT7-1-3 / Species (production host): Escherichia coli / Cellular location (production host): CYTOPLASM / Production host:  |
|---|---|
| #2: Chemical | ChemComp-A80 / 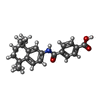  Mass: 351.439 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C22H25NO3 Mass: 351.439 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C22H25NO3 |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 8 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 8 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal grow | pH: 8 Details: IN 30% PEG4000, 0.2M LI2SO4 AND 0.1 M TRIS-HCL, PH 8.0 | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS Temperature: 277 K / Method: vapor diffusion, hanging dropDetails: drop contained equal volume of protein and reservoir solution | ||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 277 K |
|---|---|
| Diffraction source | Wavelength: 1.5418 |
| Detector | Type: RIGAKU / Detector: IMAGE PLATE |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.8→38.3 Å / Num. obs: 10128 / % possible obs: 97.6 % / Redundancy: 3.7 % / Biso Wilson estimate: 24 Å2 / Rmerge(I) obs: 0.086 / Net I/σ(I): 20.2 |
| Reflection shell | Resolution: 2.8→2.95 Å / Redundancy: 3.8 % / Rmerge(I) obs: 0.26 / Mean I/σ(I) obs: 5.2 / % possible all: 95.7 |
| Reflection | *PLUS Lowest resolution: 38 Å |
| Reflection shell | *PLUS % possible obs: 95.7 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1CBR Resolution: 2.8→30 Å / Rfactor Rfree error: 0.009 / Data cutoff high rms absF: 1543984.01 / Isotropic thermal model: GROUP / Cross valid method: THROUGHOUT / σ(F): 0 Details: BULK SOLVENT MODEL USED THE STRUCTURE WAS REFINED WITH NCS CONSTRAINTS IN CNS (I.E., THE TWO MOLECULES ARE IDENTICAL).
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 37.7 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.8→30 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | NCS model details: CONSTR | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.8→2.98 Å / Rfactor Rfree error: 0.032 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 0.3 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Num. reflection obs: 9305 / Rfactor Rfree: 0.266 / Rfactor Rwork: 0.228 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
