 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2bn2: CRYSTAL STRUCTURE OF BOVINE NEUROPHYSIN II COMPLEXED WITH THE VAS... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2bn2 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF BOVINE NEUROPHYSIN II COMPLEXED WITH THE VASOPRESSIN ANALOGUE PHE-TYR AMIDE | |||||||||
 Components Components | NEUROPHYSIN II | |||||||||
 Keywords Keywords | HORMONE / HORMONE PACKAGING / TRANSPORT / PROTEIN-PEPTIDE COMPLEX | |||||||||
| Function / homology |  Function and homology information Function and homology informationV1A vasopressin receptor binding / neurohypophyseal hormone activity / maternal aggressive behavior / vasoconstriction / neuropeptide hormone activity / positive regulation of systemic arterial blood pressure / positive regulation of vasoconstriction / secretory granule / intracellular signal transduction / : Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method |  X-RAY DIFFRACTION / SAS / Resolution: 2.8 Å X-RAY DIFFRACTION / SAS / Resolution: 2.8 Å | |||||||||
 Authors Authors | Rose, J.P. / Wang, B.C. | |||||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 1991 Title: Crystal structure of a bovine neurophysin II dipeptide complex at 2.8 A determined from the single-wavelength anomalous scattering signal of an incorporated iodine atom. Authors: Chen, L.Q. / Rose, J.P. / Breslow, E. / Yang, D. / Chang, W.R. / Furey Jr., W.F. / Sax, M. / Wang, B.C. #1: Journal: Nat.Struct.Biol. / Year: 1996Title: Crystal Structure of the Neurophysin-Oxytocin Complex Authors: Rose, J.P. / Wu, C.K. / Hsiao, C.D. / Breslow, E. / Wang, B.C. #2: Journal: Eur.J.Biochem. / Year: 1988Title: Crystals of Modified Bovine Neurophysin II Authors: Rose, J.P. / Yang, D. / Yoo, C.S. / Sax, M. / Breslow, E. / Wang, B.C. #3: Journal: J.Mol.Biol. / Year: 1979Title: Crystals of a Bovine Neurophysin II-Dipeptide Amide Complex Authors: Yoo, C.S. / Wang, B.C. / Sax, M. / Breslow, E. | |||||||||
| History |
| |||||||||
| Remark 650 | HELIX DETERMINATION METHOD: KSDSSP | |||||||||
| Remark 700 | SHEET DETERMINATION METHOD: KSDSSP |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2bn2.cif.gz | 70.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2bn2.ent.gz | 53.8 KB | Display | PDB format |
| PDBx/mmJSON format | 2bn2.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bn/2bn2ftp://data.pdbj.org/pub/pdb/validation_reports/bn/2bn2 | HTTPS FTP |
|---|
-Related structure data
| Related structure data | |
|---|---|
| Similar structure data |


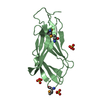








-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||||||
| 2 | 
| ||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper:
|
-Components
| #1: Protein | Mass: 9890.252 Da / Num. of mol.: 4 / Source method: isolated from a natural source / Source: (natural) #2: Chemical | ChemComp-PHE / 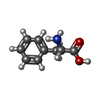  Type: L-peptide linking / Mass: 165.189 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C9H11NO2 Type: L-peptide linking / Mass: 165.189 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C9H11NO2#3: Chemical | ChemComp-TYR / 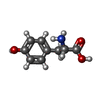  Type: L-peptide linking / Mass: 181.189 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C9H11NO3 Type: L-peptide linking / Mass: 181.189 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C9H11NO3Has protein modification | Y | Nonpolymer details | RESIDUES 98 AND 99 WITH CHAIN IDS A, C, E AND G FORM THE PHENYLALANINE-TYROSINE AMIDE DIPEPTIDE ...RESIDUES 98 AND 99 WITH CHAIN IDS A, C, E AND G FORM THE PHENYLALAN | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.82 Å3/Da / Density % sol: 56 % Description: SAS DATA WERE COLLECTED IN-HOUSE. THE ANOMALOUS SCATTERER WAS PARA IODO-PHENYLALANINE-TYROSINE AMIDE WHICH WAS BOUND IN THE HORMONE BINDING SITE. |
|---|---|
| Crystal grow | pH: 6.8 Details: 5 MG OF PROTEIN WAS DISSOLVED IN 0.5 ML OF WATER, 0.5 MG OF PHENYLALANINE-TYROSINE AMIDE AND 20 MICRO LITERS OF SATURATED AMMONIUM SULPHATE SOLUTION WERE ADDED. THE PH OF THE SOLUTION WAS ADJUSTED TO 6.8 |
| Crystal grow | *PLUS pH: 7.5 / Method: unknown |
| Components of the solutions | *PLUS Common name: ammonium sulfate |
-Data collection
| Diffraction | Mean temperature: 289 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RUH2R / Wavelength: 1.5418 |
| Detector | Type: SIEMENS / Detector: AREA DETECTOR / Date: Sep 13, 1989 / Details: SUPPER MIRRORS |
| Radiation | Monochromator: NI FILTER / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Highest resolution: 2.64 Å / Num. obs: 24898 / % possible obs: 83.6 % / Observed criterion σ(I): -3 / Redundancy: 17 % / Biso Wilson estimate: 60.2 Å2 / Rsym value: 0.0404 / Net I/σ(I): 17.05 |
| Reflection shell | Resolution: 2.64→2.74 Å / Mean I/σ(I) obs: 3.12 / Rsym value: 0.1799 / % possible all: 43.45 |
| Reflection | *PLUS Highest resolution: 2.8 Å / Num. obs: 12840 / Num. measured all: 58983 / Rmerge(I) obs: 0.0577 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SAS / Resolution: 2.8→8 Å / Rfactor Rfree error: 0.008 / Data cutoff high absF: 10000000 / Data cutoff low absF: 0.001 / Isotropic thermal model: OVERALL / Cross valid method: THROUGHOUT / σ(F): 2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 23.7 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.8→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.8→2.97 Å / Rfactor Rfree error: 0.024 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.843 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Num. reflection obs: 10729 / Rfactor obs: 0.225 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor obs: 0.298 |
