 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1ynd: Structure of human cyclophilin A in complex with the novel immuno... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1ynd | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of human cyclophilin A in complex with the novel immunosuppressant sanglifehrin A at 1.6A resolution | ||||||
 Components Components | Peptidyl-prolyl cis-trans isomerase A | ||||||
 Keywords Keywords | ISOMERASE / beta sandwich / cyclophilin-ligand complex / cyclosporin / rotamase | ||||||
| Function / homology |  Function and homology information Function and homology informationnegative regulation of protein K48-linked ubiquitination / regulation of apoptotic signaling pathway / cell adhesion molecule production / negative regulation of viral life cycle / lipid droplet organization / heparan sulfate binding / regulation of viral genome replication / virion binding / leukocyte chemotaxis / endothelial cell activation ...negative regulation of protein K48-linked ubiquitination / regulation of apoptotic signaling pathway / cell adhesion molecule production / negative regulation of viral life cycle / lipid droplet organization / heparan sulfate binding / regulation of viral genome replication / virion binding / leukocyte chemotaxis / endothelial cell activation / negative regulation of stress-activated MAPK cascade / Basigin interactions / cyclosporin A binding / Minus-strand DNA synthesis / Plus-strand DNA synthesis / Uncoating of the HIV Virion / Early Phase of HIV Life Cycle / Integration of provirus / APOBEC3G mediated resistance to HIV-1 infection / viral release from host cell / protein peptidyl-prolyl isomerization / Calcineurin activates NFAT / Binding and entry of HIV virion / positive regulation of viral genome replication / negative regulation of oxidative stress-induced intrinsic apoptotic signaling pathway / : / neutrophil chemotaxis / activation of protein kinase B activity / Gene and protein expression by JAK-STAT signaling after Interleukin-12 stimulation / negative regulation of protein phosphorylation / peptidylprolyl isomerase / positive regulation of protein secretion / peptidyl-prolyl cis-trans isomerase activity / negative regulation of protein kinase activity / Assembly Of The HIV Virion / Budding and maturation of HIV virion / neuron differentiation / platelet activation / platelet aggregation / SARS-CoV-1 activates/modulates innate immune responses / unfolded protein binding / integrin binding / protein folding / Platelet degranulation / positive regulation of NF-kappaB transcription factor activity / cellular response to oxidative stress / secretory granule lumen / vesicle / ficolin-1-rich granule lumen / positive regulation of MAPK cascade / positive regulation of protein phosphorylation / focal adhesion / Neutrophil degranulation / apoptotic process / protein-containing complex / RNA binding / extracellular space / extracellular exosome / extracellular region / membrane / nucleus / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.6 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.6 Å | ||||||
 Authors Authors | Kallen, J. / Sedrani, R. / Zenke, G. / Wagner, J. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2005 Title: Structure of human cyclophilin A in complex with the novel immunosuppressant sanglifehrin A at 1.6 A resolution. Authors: Kallen, J. / Sedrani, R. / Zenke, G. / Wagner, J. #1: Journal: J.Am.Chem.Soc. / Year: 2003Title: Sanglifehrin-Cyclophilin Interaction: Degradation Work, Synthetic Macrocyclic Analogues, X-ray Crystal Structure, and Binding Data Authors: Sedrani, R. / Kallen, J. / Cabrejas, L.M. / Papageorgiou, C.D. / Senia, F. / Rohrbach, S. / Wagner, D. / Thai, B. / Jutzi-Erne, A.M. / France, J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1ynd.cif.gz | 87.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1ynd.ent.gz | 66.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1ynd.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 1ynd_validation.pdf.gz | 960 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 1ynd_full_validation.pdf.gz | 965.4 KB | Display | |
| Data in XML | 1ynd_validation.xml.gz | 20 KB | Display | |
| Data in CIF | 1ynd_validation.cif.gz | 28.1 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/yn/1yndftp://data.pdbj.org/pub/pdb/validation_reports/yn/1ynd | HTTPS FTP |
-Related structure data
| Related structure data |  1cwaS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj











- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 18036.504 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: PPIA, CYPA / Plasmid: modified pkk 223-2 / Species (production host): Escherichia coliProduction host:  Strain (production host): W3110 / References: UniProt: P62937, peptidylprolyl isomerase #2: Chemical | 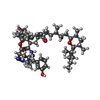  Mass: 1090.390 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C60H91N5O13 Mass: 1090.390 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C60H91N5O13#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 362 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 362 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.2 Å3/Da / Density % sol: 42.6 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 6.5 Details: Mg acetate, Na cacodylate, PEG4000, pH 6.5, VAPOR DIFFUSION, HANGING DROP, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 293 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: BM1A / Wavelength: 0.873 Å / Beamline: BM1A / Wavelength: 0.873 Å |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Jan 18, 1997 |
| Radiation | Monochromator: Si 111 CHANNEL / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.873 Å / Relative weight: 1 |
| Reflection | Resolution: 1.6→15 Å / Num. obs: 41593 / % possible obs: 99.2 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 4.3 % / Biso Wilson estimate: 14.4 Å2 / Rsym value: 0.075 / Net I/σ(I): 16.4 |
| Reflection shell | Resolution: 1.6→1.66 Å / Redundancy: 4.2 % / Mean I/σ(I) obs: 1.9 / Num. unique all: 4102 / Rsym value: 0.456 / % possible all: 100 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1CWA Resolution: 1.6→15 Å / Cor.coef. Fo:Fc: 0.966 / Cor.coef. Fo:Fc free: 0.96 / SU B: 2.205 / SU ML: 0.077 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.089 / ESU R Free: 0.084 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 14.282 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.6→15 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.6→1.642 Å / Total num. of bins used: 20
|
