 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1v2g: The L109P mutant of E. coli Thioesterase I/Protease I/Lysophospho... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1v2g | ||||||
|---|---|---|---|---|---|---|---|
| Title | The L109P mutant of E. coli Thioesterase I/Protease I/Lysophospholipase L1 (TAP) in complexed with octanoic acid | ||||||
 Components Components | Acyl-CoA thioesterase I | ||||||
 Keywords Keywords | HYDROLASE / SGNH-hydrolase fold | ||||||
| Function / homology |  Function and homology information Function and homology information: / : / : / : / B-type glycerophospholipase activity / : / arylesterase / lysophospholipase / palmitoyl-CoA hydrolase / long-chain fatty acyl-CoA hydrolase activity ...: / : / : / : / B-type glycerophospholipase activity / : / arylesterase / lysophospholipase / palmitoyl-CoA hydrolase / long-chain fatty acyl-CoA hydrolase activity / oleoyl-[acyl-carrier-protein] hydrolase / fatty acyl-[ACP] hydrolase activity / phosphatidylcholine lysophospholipase A1 activity / Hydrolases; Acting on peptide bonds (peptidases); Serine endopeptidases / arylesterase activity / lipid metabolic process / peptidase activity / outer membrane-bounded periplasmic space / periplasmic space / proteolysis / identical protein binding Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å | ||||||
 Authors Authors | Lo, Y.-C. / Lin, S.-C. / Liaw, Y.-C. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2005 Title: Substrate specificities of Escherichia coli thioesterase I/protease I/lysophospholipase L1 are governed by its switch loop movement Authors: Lo, Y.-C. / Lin, S.-C. / Shaw, J.-F. / Liaw, Y.-C. #1: Journal: J.Mol.Biol. / Year: 2003Title: Crystal structure of Escherichia coli thioesterase I/protease I/lysophospholipase L1: consensus sequence blocks constitute the catalytic center of SGNH-hydrolases through a conserved hydrogen bond network Authors: Lo, Y.-C. / Lin, S.-C. / Shaw, J.-F. / Liaw, Y.-C. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1v2g.cif.gz | 52.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1v2g.ent.gz | 36.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1v2g.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/v2/1v2gftp://data.pdbj.org/pub/pdb/validation_reports/v2/1v2g | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1u8uC  1jrlS  1nyv S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |












-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 21545.371 Da / Num. of mol.: 1 / Mutation: L109P Source method: isolated from a genetically manipulated source Source: (gene. exp.) References: UniProt: P29679, UniProt: P0ADA1*PLUS, lysophospholipase, Hydrolases; Acting on ester bonds; Thioester hydrolases |
|---|---|
| #2: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 |
| #3: Chemical | ChemComp-IMD /   Mass: 69.085 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H5N2 Mass: 69.085 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H5N2 |
| #4: Chemical | ChemComp-OCA / 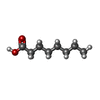  Mass: 144.211 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H16O2 Mass: 144.211 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H16O2 |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 107 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 107 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.49 Å3/Da / Density % sol: 50.18 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 6.5 Details: 2-[N-morpholino]ethanesulfonic acid, PEGMME5K, Ammonium sulfate, pH 6.5, VAPOR DIFFUSION, HANGING DROP, temperature 298K |
-Data collection
| Diffraction | Mean temperature: 133 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSRRC  / Beamline: BL17B2 / Wavelength: 1.1272 Å / Beamline: BL17B2 / Wavelength: 1.1272 Å |
| Detector | Type: MAC Science DIP-2030 / Detector: IMAGE PLATE / Date: May 30, 2000 |
| Radiation | Monochromator: Si111 Channel / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.1272 Å / Relative weight: 1 |
| Reflection | Resolution: 2→24.81 Å / Num. all: 15163 / Num. obs: 14724 / % possible obs: 97.1 % / Observed criterion σ(F): 0 / Redundancy: 9.2 % / Biso Wilson estimate: 32.7 Å2 / Limit h max: 24 / Limit h min: 0 / Limit k max: 17 / Limit k min: 0 / Limit l max: 85 / Limit l min: 0 / Observed criterion F max: 1110446.57 / Observed criterion F min: 0.77 / Rmerge(I) obs: 0.056 / Net I/σ(I): 12.1 |
| Reflection shell | Resolution: 2→2.03 Å / Rmerge(I) obs: 0.458 / Mean I/σ(I) obs: 3.2 / % possible all: 97.8 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entry 1JRL Resolution: 2→24.81 Å / Rfactor Rfree error: 0.007 / Occupancy max: 1 / Occupancy min: 1 / Isotropic thermal model: anisotropic / Cross valid method: THROUGHOUT / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: CNS bulk solvent model used / Bsol: 54.3254 Å2 / ksol: 0.338479 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 118.63 Å2 / Biso mean: 51.16 Å2 / Biso min: 27.53 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→24.81 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
|
