Journal: J.Biol.Chem. / Year: 2004 Title: Structural Determinants of Substrate Specificity in Family 1 Beta-Glucosidases: Novel Insights from the Crystal Structure of Sorghum Dhurrinase-1, a Plant Beta-Glucosidase with Strict ...Title: Structural Determinants of Substrate Specificity in Family 1 Beta-Glucosidases: Novel Insights from the Crystal Structure of Sorghum Dhurrinase-1, a Plant Beta-Glucosidase with Strict Specificity, in Complex with its Natural Substrate Authors: Verdoucq, L. / Moriniere, J. / Bevan, D.R. / Esen, A. / Vasella, A. / Henrissat, B. / Czjzek, M.
SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AA" IN EACH CHAIN ON SHEET RECORDS BELOW ... SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AA" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 9-STRANDED BARREL THIS IS REPRESENTED BY A 10-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL.
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads












 PDBj
PDBj



 Assembly
Assembly


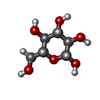
 Type: D-saccharide, beta linking / Mass: 180.156 Da / Num. of mol.: 1
Type: D-saccharide, beta linking / Mass: 180.156 Da / Num. of mol.: 1
 Mass: 41.052 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H3N
Mass: 41.052 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H3N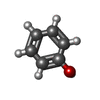
 Mass: 94.111 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H6O
Mass: 94.111 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H6O Mass: 18.015 Da / Num. of mol.: 600 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 600 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: ID14-2 / Wavelength: 0.934
/ Beamline: ID14-2 / Wavelength: 0.934  Processing
Processing