| Entry | Database: PDB / ID: 1upr
|
|---|
| Title | Crystal structure of the PEPP1 pleckstrin homology domain in complex with Inositol 1,3,4,5-tetrakisphosphate |
|---|
 Components Components | PLECKSTRIN HOMOLOGY DOMAIN-CONTAINING FAMILY A MEMBER 4 |
|---|
 Keywords Keywords | SIGNALING PROTEIN / SIGNAL TRANSDUCTION / PHOSPHOINOSITIDE BINDING DOMAIN |
|---|
| Function / homology |  Function and homology information Function and homology information
positive regulation of Wnt signaling pathway, planar cell polarity pathway / phosphatidylinositol biosynthetic process / phosphatidylinositol-3-phosphate binding / phosphatidylinositol-3,4-bisphosphate binding / phosphatidylinositol-3,5-bisphosphate binding / extrinsic component of cytoplasmic side of plasma membrane / Synthesis of PIPs at the plasma membrane / phosphatidylinositol-4,5-bisphosphate binding / positive regulation of canonical Wnt signaling pathway / identical protein binding ...positive regulation of Wnt signaling pathway, planar cell polarity pathway / phosphatidylinositol biosynthetic process / phosphatidylinositol-3-phosphate binding / phosphatidylinositol-3,4-bisphosphate binding / phosphatidylinositol-3,5-bisphosphate binding / extrinsic component of cytoplasmic side of plasma membrane / Synthesis of PIPs at the plasma membrane / phosphatidylinositol-4,5-bisphosphate binding / positive regulation of canonical Wnt signaling pathway / identical protein binding / plasma membrane / cytoplasmSimilarity search - Function PKHA4-7, PH domain / : / PH domain-containing proteins, TBCA domain / Pleckstrin-homology domain (PH domain)/Phosphotyrosine-binding domain (PTB) / PH-domain like / PH domain / PH domain profile. / Pleckstrin homology domain. / Pleckstrin homology domain / PH-like domain superfamily ...PKHA4-7, PH domain / : / PH domain-containing proteins, TBCA domain / Pleckstrin-homology domain (PH domain)/Phosphotyrosine-binding domain (PTB) / PH-domain like / PH domain / PH domain profile. / Pleckstrin homology domain. / Pleckstrin homology domain / PH-like domain superfamily / Roll / Mainly BetaSimilarity search - Domain/homology |
|---|
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) |
|---|
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.27 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.27 Å |
|---|
 Authors Authors | Milburn, C.C. / Komander, D. / Deak, M. / Alessi, D.R. / Van Aalten, D.M.F. |
|---|
 Citation Citation | Journal: To be Published
Title: Crystal Structure of the Pleckstrin Homology Domain of Pepp1
Authors: Milburn, C.C. / Komander, D. / Deak, M. / Alessi, D.R. / Van Aalten, D.M.F. |
|---|
| History | | Deposition | Oct 10, 2003 | Deposition site: PDBE / Processing site: PDBE |
|---|
| Revision 1.0 | Oct 20, 2004 | Provider: repository / Type: Initial release |
|---|
| Revision 1.1 | May 16, 2012 | Group: Database references / Derived calculations ...Database references / Derived calculations / Non-polymer description / Other / Refinement description / Structure summary / Version format compliance |
|---|
| Revision 1.2 | Dec 7, 2016 | Group: Database references |
|---|
| Revision 1.3 | Dec 13, 2023 | Group: Data collection / Database references ...Data collection / Database references / Derived calculations / Other / Refinement description
Category: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / pdbx_database_status / pdbx_initial_refinement_model / struct_site
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession ..._database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_database_status.status_code_sf / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id |
|---|
|
|---|
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj
 Assembly
Assembly

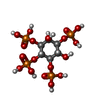
 Mass: 500.075 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H16O18P4
Mass: 500.075 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H16O18P4 Mass: 18.015 Da / Num. of mol.: 29 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 29 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: PX14.2 / Wavelength: 0.97668
/ Beamline: PX14.2 / Wavelength: 0.97668  Processing
Processing