 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1trr: TANDEM BINDING IN CRYSTALS OF A TRP REPRESSOR/OPERATOR HALF-SITE ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1trr | ||||||
|---|---|---|---|---|---|---|---|
| Title | TANDEM BINDING IN CRYSTALS OF A TRP REPRESSOR/OPERATOR HALF-SITE COMPLEX | ||||||
 Components Components |
| ||||||
 Keywords Keywords | TRANSCRIPTION/DNA / PROTEIN-DNA COMPLEX / TRANSCRIPTION-DNA COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationsequence-specific DNA binding / DNA-binding transcription factor activity / negative regulation of DNA-templated transcription / regulation of DNA-templated transcription / DNA binding / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2.4 Å X-RAY DIFFRACTION / Resolution: 2.4 Å | ||||||
 Authors Authors | Lawson, C.L. / Carey, J. | ||||||
 Citation Citation | Journal: Nature / Year: 1993 Title: Tandem binding in crystals of a trp repressor/operator half-site complex. Authors: Lawson, C.L. / Carey, J. #1: Journal: J.Mol.Biol. / Year: 1993Title: Cocrystals of E. Coli Trp Repressor Bound to an Alternative Operator DNA Sequence Authors: Carey, J. / Combatti, N. / Lewis, D.E.A. / Lawson, C.L. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1trr.cif.gz | 194.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1trr.ent.gz | 143.6 KB | Display | PDB format |
| PDBx/mmJSON format | 1trr.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/tr/1trrftp://data.pdbj.org/pub/pdb/validation_reports/tr/1trr | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj







































- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||
| 2 | 
| ||||||||||||
| Unit cell |
| ||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper:
| ||||||||||||
| Details | THE TRANSFORMATION PRESENTED ON *MTRIX 1* RECORDS BELOW WILL YIELD APPROXIMATE COORDINATES FOR CHAINS G, H, AND I WHEN APPLIED TO CHAINS A, B, AND C, RESPECTIVELY. THE TRANSFORMATION PRESENTED ON *MTRIX 2* RECORDS BELOW WILL YIELD APPROXIMATE COORDINATES FOR CHAINS D, E, F, J, K, AND L WHEN APPLIED TO CHAINS A, B, C, G, H, AND I, RESPECTIVELY. |
-Components
| #1: DNA chain | Mass: 4898.191 Da / Num. of mol.: 4 / Source method: obtained synthetically #2: Protein | Mass: 12239.919 Da / Num. of mol.: 8 / Source method: isolated from a natural source / Source: (natural) #3: Chemical | ChemComp-TRP / 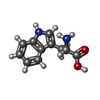  Type: L-peptide linking / Mass: 204.225 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C11H12N2O2 Type: L-peptide linking / Mass: 204.225 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C11H12N2O2#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 192 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 192 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal grow | *PLUS Temperature: 23 ℃ / pH: 4.8 / Method: vapor diffusion, hanging drop | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Components of the solutions | *PLUS
|
-Data collection
| Detector | Type: ENRAF-NONIUS FAST / Detector: DIFFRACTOMETER |
|---|---|
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Relative weight: 1 |
| Reflection | Num. all: 30065 / Num. obs: 18723 |
| Reflection | *PLUS Highest resolution: 2.4 Å / % possible obs: 90.3 % / Rmerge(I) obs: 0.05 |
- Processing
Processing
| Software | Name: X-PLOR / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Highest resolution: 2.4 Å Details: THE MODEL IS MISSING THREE CARBOXYL-TERMINAL RESIDUES FROM EACH REPRESSOR SUBUNIT (106 - 108) AND RESIDUE 1 OF EACH OLIGONUCLEOTIDE (= UNPAIRED 5' THYMIDINE). THEY ARE NOT SEEN IN ELECTRON ...Details: THE MODEL IS MISSING THREE CARBOXYL-TERMINAL RESIDUES FROM EACH REPRESSOR SUBUNIT (106 - 108) AND RESIDUE 1 OF EACH OLIGONUCLEOTIDE (= UNPAIRED 5' THYMIDINE). THEY ARE NOT SEEN IN ELECTRON DENSITY AND, THEREFORE, ARE PRESUMED TO BE DISORDERED. THE AMINO-TERMINAL ARMS (RESIDUES 2 - 16) OF EACH REPRESSOR SUBUNIT WERE MODELLED INTO FAIRLY POOR BUT NEARLY CONTINUOUS ELECTRON DENSITY. THE ARMS WERE BUILT USING THE MOST CLEARLY DEFINED RESIDUES (PRO 5, TYR 7, AND HIS 16) AS GUIDES. THE LEVEL OF UNCERTAINTY IN SPECIFIC ATOM POSITIONS OF THE ARM IS REFLECTED IN POORER STEREOCHEMISTRY OF RESIDUES 2 - 16 WHEN COMPARED TO THE REST OF THE MODEL.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Highest resolution: 2.4 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.4 Å / Rfactor obs: 0.241 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: x_angle_d / Dev ideal: 1.86 |
