 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1tro: CRYSTAL STRUCTURE OF TRP REPRESSOR OPERATOR COMPLEX AT ATOMIC RES... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1tro | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF TRP REPRESSOR OPERATOR COMPLEX AT ATOMIC RESOLUTION | ||||||
 Components Components |
| ||||||
 Keywords Keywords | TRANSCRIPTION/DNA / PROTEIN-DNA COMPLEX / DOUBLE HELIX / TRANSCRIPTION-DNA COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationsequence-specific DNA binding / DNA-binding transcription factor activity / negative regulation of DNA-templated transcription / regulation of DNA-templated transcription / DNA binding / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 1.9 Å X-RAY DIFFRACTION / Resolution: 1.9 Å | ||||||
 Authors Authors | Otwinowski, Z. / Schevitz, R.W. / Zhang, R.-G. / Lawson, C.L. / Joachimiak, A. / Marmorstein, R. / Luisi, B.F. / Sigler, P.B. | ||||||
 Citation Citation | Journal: Nature / Year: 1988 Title: Crystal structure of trp repressor/operator complex at atomic resolution. Authors: Otwinowski, Z. / Schevitz, R.W. / Zhang, R.G. / Lawson, C.L. / Joachimiak, A. / Marmorstein, R.Q. / Luisi, B.F. / Sigler, P.B. #1: Journal: J.Biol.Chem. / Year: 1987Title: Crystal of the Trp Repressor-Operator Complex Suitable for X-Ray Diffraction Analysis Authors: Joachimiak, A. / Marmorstein, R. / Schevitz, R. / Mandecki, W. / Fox, J.L. / Sigler, P.B. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1tro.cif.gz | 157 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1tro.ent.gz | 110.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1tro.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/tr/1troftp://data.pdbj.org/pub/pdb/validation_reports/tr/1tro | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj







































- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | THE NON-CRYSTALLOGRAPHIC SYMMETRY OF THE STRUCTURE IS NOT EXACT AND THE DEPARTURES FROM THE NON-CRYSTALLOGRAPHIC FOUR-FOLD SYMMETRY DO NOT SEEM TO BE FUNCTIONALLY SIGNIFICANT. TRP REPRESSOR IS A STABLE DIMERIC PROTEIN. ANY ANALYSIS OF OF THIS STRUCTURE REQUIRES THAT A FULL DIMER BE LOOKED AT ONE TIME. THE COORDINATES CONTAIN TWO DIMERS. THE COMPLEX IS MADE BY FOLLOWING CHAIN NUMBERS. COMPLEX 1: A,B,C,D,I,J; COMPLEX 2: E,F,G,H,K,L. |
-Components
| #1: DNA chain | Mass: 5818.796 Da / Num. of mol.: 4 / Source method: obtained synthetically #2: Protein | Mass: 12371.115 Da / Num. of mol.: 4 / Source method: isolated from a natural source Source: (natural) Species: Escherichia coli / Strain: W3110 / References: UniProt: P0A881 #3: Chemical | ChemComp-TRP / 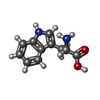  Type: L-peptide linking / Mass: 204.225 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C11H12N2O2 Type: L-peptide linking / Mass: 204.225 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C11H12N2O2#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 572 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 572 / Source method: isolated from a natural source / Formula: H2ONonpolymer details | THE REGULATORY | Sequence details | SEQUENCE ADVISORY NOTICE: DIFFERENCE BETWEEN SWISS-PROT AND PDB SEQUENCE. SWISS-PROT ENTRY NAME: ...SEQUENCE ADVISORY NOTICE: DIFFERENCE | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.3 Å3/Da / Density % sol: 46.51 % | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Method: vapor diffusion / Details: VAPOR DIFFUSION | ||||||||||||||||||||||||||||||||||||
| Components of the solutions |
| ||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: unknown | ||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
|---|---|
| Radiation wavelength | Relative weight: 1 |
| Reflection | *PLUS Highest resolution: 2.4 Å / Lowest resolution: 10 Å / Num. all: 26050 / % possible obs: 87.3 % / Rmerge(I) obs: 0.11 |
- Processing
Processing
| Software | Name: PROLSQ / Classification: refinement | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 1.9→10 Å Details: THE CRYSTALLOGRAPHIC TEMPERATURE FACTORS OF THE FIRST TWO PROTEIN AND DNA CHAINS ARE LOWER THAN THE SUBSEQUENT TWO; CONSEQUENTLY THE STRUCTURE OF THE FIRST TWO CHAINS IS MORE ACCURATELY DEFINED.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→10 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: PROLSQ / Classification: refinement | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.4 Å / Lowest resolution: 10 Å / Rfactor obs: 0.249 / Rfactor Rwork: 0.249 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
