 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1tcb: THE SEQUENCE, CRYSTAL STRUCTURE DETERMINATION AND REFINEMENT OF T... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1tcb | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | THE SEQUENCE, CRYSTAL STRUCTURE DETERMINATION AND REFINEMENT OF TWO CRYSTAL FORMS OF LIPASE B FROM CANDIDA ANTARCTICA | |||||||||
 Components Components | LIPASE | |||||||||
 Keywords Keywords | HYDROLASE(CARBOXYLIC ESTERASE) | |||||||||
| Function / homology |  Function and homology information Function and homology informationtriacylglycerol lipase / triacylglycerol lipase activity / lipid catabolic process Similarity search - Function | |||||||||
| Biological species |  Candida antarctica (fungus) Candida antarctica (fungus) | |||||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2.1 Å X-RAY DIFFRACTION / Resolution: 2.1 Å | |||||||||
 Authors Authors | Uppenberg, J. / Jones, T.A. | |||||||||
 Citation Citation | Journal: Structure / Year: 1994 Title: The sequence, crystal structure determination and refinement of two crystal forms of lipase B from Candida antarctica. Authors: Uppenberg, J. / Hansen, M.T. / Patkar, S. / Jones, T.A. #1: Journal: J.Mol.Biol. / Year: 1994Title: Crystallization and Preliminary X-Ray Studies of Lipase B from Candida Antarctica Authors: Uppenberg, J. / Patkar, S. / Bergfors, T. / Jones, T.A. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1tcb.cif.gz | 138.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1tcb.ent.gz | 106.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1tcb.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/tc/1tcbftp://data.pdbj.org/pub/pdb/validation_reports/tc/1tcb | HTTPS FTP |
|---|
-Related structure data









-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Atom site foot note | 1: CIS PROLINE - PRO A 70 / 2: CIS PROLINE - PRO A 192 / 3: CIS PROLINE - PRO B 70 / 4: CIS PROLINE - PRO B 192 | ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (0.2738, 0.9363, 0.2201), Vector: Details | THIS IS THE LOW PH STRUCTURE OF THE MONOCLINIC CRYSTAL; FORM WITH TWO MOLECULES IN THE ASYMMETRIC UNIT. THE MOLECULES ARE CALLED A AND B. THE RMS DEVIATIONS ARE 0.18 ANGSTROMS FOR C-ALPHAS AND 0.31 ANGSTROMS FOR ALL ATOMS. THE TRANSFORMATION PRESENTED ON *MTRIX* RECORDS BELOW WILL YIELD APPROXIMATE COORDINATES FOR CHAIN B WHEN APPLIED TO CHAIN A. | |
-Components
| #1: Protein | Mass: 33040.238 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Candida antarctica (fungus) / References: UniProt: P41365, triacylglycerol lipase#2: Polysaccharide | 2-acetamido-2-deoxy-beta-D-glucopyranose-(1-4)-2-acetamido-2-deoxy-beta-D-glucopyranose | Source method: isolated from a genetically manipulated source #3: Sugar | 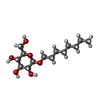  Type: D-saccharide / Mass: 292.369 Da / Num. of mol.: 2 Type: D-saccharide / Mass: 292.369 Da / Num. of mol.: 2Source method: isolated from a genetically manipulated source Formula: C14H28O6 / Comment: detergent*YM #4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 470 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 470 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | Sequence details | THE SEQUENCE HAS NOT BEEN REPORTED. IT WAS DERIVED FROM THE DNA SEQUENCE OF J.UPPENBERG ET AL., ...THE SEQUENCE HAS NOT BEEN REPORTED. IT WAS DERIVED FROM THE DNA SEQUENCE OF J.UPPENBERG ET AL., 1994, LISTED IN THE JRNL REFERENCE ABOVE. | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.24 Å3/Da / Density % sol: 45.2 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS pH: 3.6 / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Reflection | *PLUS Highest resolution: 2.1 Å / Num. obs: 31323 / % possible obs: 86 % / Num. measured all: 58582 / Rmerge(I) obs: 0.03 |
|---|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.1→7.5 Å / σ(F): 2 /
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.1→7.5 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.187 / Rfactor Rwork: 0.187 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: x_angle_d / Dev ideal: 0.9 |
