 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1qwn: GOLGI ALPHA-MANNOSIDASE II Covalent Intermediate Complex with 5-f... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1qwn | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | GOLGI ALPHA-MANNOSIDASE II Covalent Intermediate Complex with 5-fluoro-gulosyl-fluoride | |||||||||
 Components Components | Alpha-mannosidase II | |||||||||
 Keywords Keywords | HYDROLASE / N-TERMINAL ALPHA-BETA DOMAIN / THREE HELIX BUNDLE / 2 C-TERMINAL BETA BARRELS / Family 38 Glycosyl hydrolase | |||||||||
| Function / homology |  Function and homology information Function and homology informationmannosyl-oligosaccharide 1,3-1,6-alpha-mannosidase / mannosyl-oligosaccharide 1,3-1,6-alpha-mannosidase activity / rhodopsin biosynthetic process / encapsulation of foreign target / Golgi apparatus N-glycan mannose trimming / Reactions specific to the complex N-glycan synthesis pathway / mannosidase activity / alpha-mannosidase activity / mannose metabolic process / N-glycan processing ...mannosyl-oligosaccharide 1,3-1,6-alpha-mannosidase / mannosyl-oligosaccharide 1,3-1,6-alpha-mannosidase activity / rhodopsin biosynthetic process / encapsulation of foreign target / Golgi apparatus N-glycan mannose trimming / Reactions specific to the complex N-glycan synthesis pathway / mannosidase activity / alpha-mannosidase activity / mannose metabolic process / N-glycan processing / Golgi stack / carbohydrate binding / Golgi membrane / endoplasmic reticulum / metal ion binding Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.2 Å | |||||||||
 Authors Authors | Numao, S. / Kuntz, D.A. / Withers, S.G. / Rose, D.R. | |||||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2003 Title: Insights into the mechanism of Drosophila melanogaster Golgi alpha-mannosidase II through the structural analysis of covalent reaction intermediates. Authors: Numao, S. / Kuntz, D.A. / Withers, S.G. / Rose, D.R. | |||||||||
| History |
| |||||||||
| Remark 999 | SEQUENCE The E -> K conflict for residue 970 is noted in Swiss-Prot entry Q24451. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1qwn.cif.gz | 249 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1qwn.ent.gz | 194.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1qwn.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/qw/1qwnftp://data.pdbj.org/pub/pdb/validation_reports/qw/1qwn | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1qwuC  1qx1C  1htyS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |























-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 119701.617 Da / Num. of mol.: 1 / Fragment: Family 38 catalytic domain (residues 94-1108) Source method: isolated from a genetically manipulated source Source: (gene. exp.) References: UniProt: Q24451, mannosyl-oligosaccharide 1,3-1,6-alpha-mannosidase |
|---|
-Sugars , 2 types, 2 molecules 
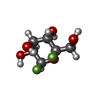
| #2: Sugar | ChemComp-NAG /  Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 1 Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 1Source method: isolated from a genetically manipulated source Formula: C8H15NO6 |
|---|---|
| #4: Sugar | ChemComp-GUL / ( Type: L-saccharide / Mass: 200.137 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H10F2O5 Type: L-saccharide / Mass: 200.137 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H10F2O5 |
-Non-polymers , 4 types, 1037 molecules 



| #3: Chemical | ChemComp-ZN /  Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn |
|---|---|
| #5: Chemical | ChemComp-TRS /  Mass: 122.143 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H12NO3 / Comment: pH buffer*YM Mass: 122.143 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H12NO3 / Comment: pH buffer*YM |
| #6: Chemical | ChemComp-MPD / ( Mass: 118.174 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H14O2 / Comment: precipitant*YM Mass: 118.174 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H14O2 / Comment: precipitant*YM |
| #7: Water | ChemComp-HOH / Mass: 18.015 Da / Num. of mol.: 1034 / Source method: isolated from a natural source / Formula: H2O |
-Details
| Has protein modification | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.2 Å3/Da / Density % sol: 44.16 % | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 7 Details: PEG 6000, MPD, Tris, pH 7, VAPOR DIFFUSION, HANGING DROP, temperature 298K | ||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 7 / Method: vapor diffusion / Details: van den Elsen, J.M., (2001) EMBO J., 20, 3008. | ||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: CHESS  / Beamline: F1 / Wavelength: 1 Å / Beamline: F1 / Wavelength: 1 Å |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Mar 24, 2002 / Details: monochromator |
| Radiation | Monochromator: Si(111) Double Crystal / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.2→30 Å / Num. all: 328509 / Num. obs: 317663 / % possible obs: 96.7 % / Observed criterion σ(F): 2.9 / Observed criterion σ(I): 2 / Redundancy: 7.5 % / Rmerge(I) obs: 0.075 / Rsym value: 0.075 / Net I/σ(I): 23.7 |
| Reflection shell | Resolution: 1.2→1.22 Å / Rmerge(I) obs: 0.429 / Mean I/σ(I) obs: 3.4 / % possible all: 91.4 |
| Reflection | *PLUS Highest resolution: 1.2 Å / Lowest resolution: 30 Å / Num. obs: 324605 / % possible obs: 98.8 % / Num. measured all: 2449245 |
| Reflection shell | *PLUS % possible obs: 91.4 % |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1HTY Resolution: 1.2→30 Å / Isotropic thermal model: isotropic / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: ENGH & HUBER
| |||||||||||||||||||||||||
| Displacement parameters | Biso mean: 12.8 Å2 | |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.2→30 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 30 Å | |||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: c_angle_deg / Dev ideal: 1.97 |
