 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1qmo: Structure of FRIL, a legume lectin that delays hematopoietic prog... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1qmo | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of FRIL, a legume lectin that delays hematopoietic progenitor maturation | ||||||
 Components Components | (MANNOSE BINDING LECTIN, FRIL) x 2 | ||||||
 Keywords Keywords | LECTIN / CROSSLINK / HEMATOPOIETIC PROGENITOR / SUGAR COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  DOLICHOS LAB LAB (hyacinth bean) DOLICHOS LAB LAB (hyacinth bean) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.5 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.5 Å | ||||||
 Authors Authors | Hamelryck, T.W. / Moore, J.G. / Chrispeels, M. / Loris, R. / Wyns, L. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2000 Title: The Role of Weak Protein-Protein Interactions in Multivalent Lectin-Carbohydrate Binding: Crystal Structure of Cross-Linked Fril Authors: Hamelryck, T.W. / Moore, J.G. / Chrispeels, M.J. / Loris, R. / Wyns, L. #1: Journal: Proc.Natl.Acad.Sci.USA / Year: 1999 Title: Cdna Cloning of Fril, a Lectin from Dolichos Lablab, that Preserves Hematopoietic Progenitors in Suspension Culture. Authors: Colucci, G. / Moore, J.G. / Feldman, M. / Chrispeels, M.J. #2: Journal: Glycobiology / Year: 1999 Title: Purification and Characterization of Dolichos Lablab Lectin Authors: Mo, H. / Meah, Y. / Moore, J.G. / Goldstein, I.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1qmo.cif.gz | 186.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1qmo.ent.gz | 150.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1qmo.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/qm/1qmoftp://data.pdbj.org/pub/pdb/validation_reports/qm/1qmo | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||||||
| Unit cell |
| ||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper:
| ||||||||||||||||
| Details | THE BIOMOLECULE CONSISTS OF A TETRAMER OF A HETERO-DIMER.THE HETERO DIMER APPEARS TO BE A RESULT OF POST-TRANSLATIONALPROCESSING OF THE SINGLE GENE PRODUCT. |
-Components
| #1: Protein | Mass: 12212.475 Da / Num. of mol.: 4 / Fragment: ALPHA CHAIN RESIDUES 1 TO 113 / Source method: isolated from a natural source / Source: (natural) DOLICHOS LAB LAB (hyacinth bean) / Organ: SEED / References: GenBank: 4204466, UniProt: Q9ZTA9*PLUS#2: Protein | Mass: 14988.616 Da / Num. of mol.: 4 / Fragment: BETA CHAIN RESIDUES 132 TO 264 / Source method: isolated from a natural source / Source: (natural) DOLICHOS LAB LAB (hyacinth bean) / Organ: SEED / References: GenBank: 4204466, UniProt: Q9ZTA9*PLUS#3: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ca#4: Chemical | ChemComp-MN /   Mass: 54.938 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mn Mass: 54.938 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mn#5: Sugar | ChemComp-MAN / 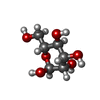  Type: D-saccharide, alpha linking / Mass: 180.156 Da / Num. of mol.: 4 Type: D-saccharide, alpha linking / Mass: 180.156 Da / Num. of mol.: 4Source method: isolated from a genetically manipulated source Formula: C6H12O6 Has protein modification | N | Sequence details | THE HETERO-DIMER, CHAINS A,E ARE THE PRODUCT OF A SINGLE GENE AND IS LIKELY DUE TO PROTEOLYTI | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 6 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 5.12 Å3/Da / Density % sol: 70 % | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 6.5 Details: METHOD 'HANING DROP BOTTOM', 20 % PEG 8000, 0.1 M NACACODYLATE, PH 6.5, 0.2 M (NH4)2SO4, WITH A 5 MICROLITER PROTEIN SOLUTION DROP, (4.3 MG/ML)+ 5 MICROLITER BOTTOM SOLUTION + 1 MICROLITER ...Details: METHOD 'HANING DROP BOTTOM', 20 % PEG 8000, 0.1 M NACACODYLATE, PH 6.5, 0.2 M (NH4)2SO4, WITH A 5 MICROLITER PROTEIN SOLUTION DROP, (4.3 MG/ML)+ 5 MICROLITER BOTTOM SOLUTION + 1 MICROLITER TRIMANNOSIDE SOLUTION (90 MM) | ||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 287 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: BW7A / Wavelength: 0.92 / Beamline: BW7A / Wavelength: 0.92 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.92 Å / Relative weight: 1 |
| Reflection | Resolution: 3.5→20 Å / Num. obs: 333806 / % possible obs: 99.4 % / Redundancy: 12.4 % / Rmerge(I) obs: 0.16 / Net I/σ(I): 11.3 |
| Reflection shell | Resolution: 3.5→3.62 Å / Mean I/σ(I) obs: 4.8 |
| Reflection | *PLUS Num. obs: 26919 / Num. measured all: 257389 / Rmerge(I) obs: 0.159 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: MODEL Resolution: 3.5→20 Å / Rfactor Rfree error: 0.005 / Data cutoff high absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 Details: NO DENSITY FOR RESIDUES AFTER POSITION 248. GAP BETWEEN ASN 113 AND SER 132, LIKELY DUE TO PROTEOLYTICAL PROCESSING
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 10 Å2 / ksol: 0.168158 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 49.2 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.5→20 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 3.5→3.72 Å / Rfactor Rfree error: 0.015 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 0.4 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
