 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1qcu: CRYSTAL STRUCTURE OF AN 18 BASE PAIR COPY CONTROL RELATED RNA DUPLEX -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1qcu | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF AN 18 BASE PAIR COPY CONTROL RELATED RNA DUPLEX | ||||||
 Components Components |
| ||||||
 Keywords Keywords | RNA / A-RNA STRUCTURE / RIBONUCLEIC ACID | ||||||
| Function / homology | AMMONIUM ION / RNA / RNA (> 10) Function and homology information Function and homology information | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.2 Å | ||||||
 Authors Authors | Klosterman, P.S. / Shah, S.A. / Steitz, T.A. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 1999 Title: Crystal structures of two plasmid copy control related RNA duplexes: An 18 base pair duplex at 1.20 A resolution and a 19 base pair duplex at 1.55 A resolution. Authors: Klosterman, P.S. / Shah, S.A. / Steitz, T.A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1qcu.cif.gz | 74.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1qcu.ent.gz | 59 KB | Display | PDB format |
| PDBx/mmJSON format | 1qcu.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/qc/1qcuftp://data.pdbj.org/pub/pdb/validation_reports/qc/1qcu | HTTPS FTP |
|---|
-Related structure data


-Links
 PDBj
PDBj






























- Assembly
Assembly
| Deposited unit | 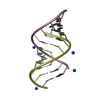
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||||||||||||||||||||||||||
| 2 | 
| |||||||||||||||||||||||||||||||||
| Unit cell |
| |||||||||||||||||||||||||||||||||
| Components on special symmetry positions |
|
-Components
| #1: RNA chain | Mass: 3752.306 Da / Num. of mol.: 2 / Source method: obtained synthetically #2: RNA chain | Mass: 3312.042 Da / Num. of mol.: 2 / Source method: obtained synthetically #3: Chemical | ChemComp-NH4 / 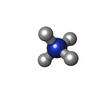  Mass: 18.038 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: H4N Mass: 18.038 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: H4N#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 195 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 195 / Source method: isolated from a natural source / Formula: H2OCompound details | THE CRYSTALS EXHIBIT 36-FOLD STATIC DISORDER, SUCH THAT THERE IS ONE CONFORMATION OF THE SUGAR- ...THE CRYSTALS EXHIBIT 36-FOLD STATIC DISORDER, SUCH THAT THERE IS ONE CONFORMATI | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 2 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.4 Å3/Da / Density % sol: 49.5 % | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 6.5 Details: (NH4)2SO4, SODIUM CACODYLATE, MGCL2, ROP PROTEIN, pH 6.5, VAPOR DIFFUSION, HANGING DROP, temperature 293K | ||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions |
| ||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 20 ℃ / Method: vapor diffusion | ||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction |
| ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source |
| ||||||||||||||||||
| Detector |
| ||||||||||||||||||
| Radiation |
| ||||||||||||||||||
| Radiation wavelength |
| ||||||||||||||||||
| Reflection | Resolution: 1.2→30 Å / Num. all: 19666 / Num. obs: 18266 / % possible obs: 92.5 % / Observed criterion σ(F): -3 / Observed criterion σ(I): -3 / Redundancy: 4.2 % / Biso Wilson estimate: 21.7 Å2 / Rmerge(I) obs: 0.073 / Net I/σ(I): 31.8 | ||||||||||||||||||
| Reflection shell | Resolution: 1.2→1.24 Å / Redundancy: 2.8 % / Rmerge(I) obs: 0.274 / Mean I/σ(I) obs: 5.2 / % possible all: 77.1 | ||||||||||||||||||
| Reflection | *PLUS % possible obs: 93 % | ||||||||||||||||||
| Reflection shell | *PLUS % possible obs: 77 % |

- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: A-RNA Resolution: 1.2→30 Å / Num. parameters: 7977 / Num. restraintsaints: 21649 / Cross valid method: FREE R / σ(F): 0 / σ(I): 0 StereochEM target val spec case: USED THE AVERAGE OF GUA C6 O6, ADE C6 N6 BOND LENGTHS AS THE C6 O6 BOND LENGTH Stereochemistry target values: Parkinson et al. Details: ANISOTROPIC REFINEMENT REDUCED FREE R (NO CUTOFF) BY 6.5% THE MOLECULE CRYSTALLIZED IN SPACE GROUP P321. HOWEVER THE REFINEMENT WAS CARRIED OUT IN THE LOWER SPACE GROUP P3. THE COORDINATES ...Details: ANISOTROPIC REFINEMENT REDUCED FREE R (NO CUTOFF) BY 6.5% THE MOLECULE CRYSTALLIZED IN SPACE GROUP P321. HOWEVER THE REFINEMENT WAS CARRIED OUT IN THE LOWER SPACE GROUP P3. THE COORDINATES DEPOSITED ARE FOR THE SPACE GROUP P3.
| |||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: MOEWS & KRETSINGER, J.MOL.BIOL., 91 201-228 (1973) | |||||||||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 22 / Occupancy sum hydrogen: 227 / Occupancy sum non hydrogen: 578 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.2→30 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||
| Software | *PLUS Name: SHELXL-97 / Classification: refinement | |||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor all: 0.202 / Rfactor obs: 0.116 / Rfactor Rfree: 0.15 | |||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
