 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1p43: REVERSE PROTONATION IS THE KEY TO GENERAL ACID-BASE CATALYSIS IN ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1p43 | ||||||
|---|---|---|---|---|---|---|---|
| Title | REVERSE PROTONATION IS THE KEY TO GENERAL ACID-BASE CATALYSIS IN ENOLASE | ||||||
 Components Components | Enolase 1 | ||||||
 Keywords Keywords | LYASE / Beta Barrel | ||||||
| Function / homology |  Function and homology information Function and homology informationGluconeogenesis / regulation of vacuole fusion, non-autophagic / Glycolysis / melatonin binding / phosphopyruvate hydratase / phosphopyruvate hydratase complex / phosphopyruvate hydratase activity / fungal-type vacuole / glycolytic process / magnesium ion binding ...Gluconeogenesis / regulation of vacuole fusion, non-autophagic / Glycolysis / melatonin binding / phosphopyruvate hydratase / phosphopyruvate hydratase complex / phosphopyruvate hydratase activity / fungal-type vacuole / glycolytic process / magnesium ion binding / mitochondrion / plasma membrane / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.8 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.8 Å | ||||||
 Authors Authors | Sims, P.A. / Larsen, T.M. / Poyner, R.R. / Cleland, W.W. / Reed, G.H. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2003 Title: Reverse protonation is the key to general acid-base catalysis in enolase Authors: Sims, P.A. / Larsen, T.M. / Poyner, R.R. / Cleland, W.W. / Reed, G.H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1p43.cif.gz | 193.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1p43.ent.gz | 151.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1p43.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/p4/1p43ftp://data.pdbj.org/pub/pdb/validation_reports/p4/1p43 | HTTPS FTP |
|---|
-Related structure data















-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
| |||||||||
| Details | The biological assembly is a dimer. The asymmetric unit contains one dimer. |
-Components
| #1: Protein | Mass: 46731.812 Da / Num. of mol.: 2 / Mutation: E168Q Source method: isolated from a genetically manipulated source Source: (gene. exp.) Gene: ENO1 OR ENOA OR HSP48 OR YGR254W OR G9160 / Production host:  #2: Chemical | ChemComp-MG /   Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg#3: Chemical | 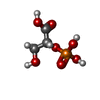  Mass: 186.057 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H7O7P Mass: 186.057 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H7O7P#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 867 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 867 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.39 Å3/Da / Density % sol: 48.56 % | |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / Method: batch / pH: 8 Details: PEG 8000, potassium chloride, HEPPS, pH 8.0, Batch, temperature 293K | |||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: batch method | |||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 110 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: ENRAF-NONIUS FR591 / Wavelength: 1.5418 Å |
| Detector | Type: BRUKER PROTEUM / Detector: CCD / Date: Oct 23, 2002 |
| Radiation | Monochromator: Montel optics / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.8→30 Å / Num. all: 83994 / Num. obs: 83994 / % possible obs: 99 % / Observed criterion σ(F): 1 / Observed criterion σ(I): 1 |
| Reflection shell | Highest resolution: 1.8 Å / % possible all: 99 |
| Reflection | *PLUS Highest resolution: 1.8 Å / Lowest resolution: 30 Å / % possible obs: 99 % / Num. measured all: 303129 / Rmerge(I) obs: 0.047 |
| Reflection shell | *PLUS Lowest resolution: 1.88 Å / % possible obs: 99 % / Rmerge(I) obs: 0.217 |
- Processing
Processing
| Software |
| |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.8→30 Å / Cross valid method: THROUGHOUT / σ(F): 1.8 / Stereochemistry target values: Engh & Huber
| |||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.8→30 Å
| |||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.8 Å / Lowest resolution: 30 Å / % reflection Rfree: 5 % | |||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||
| Refine LS restraints | *PLUS Type: c_bond_d / Dev ideal: 0.005 |
