 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 1onx | ||||||
|---|---|---|---|---|---|---|---|
| タイトル | Crystal structure of isoaspartyl dipeptidase from escherichia coli complexed with aspartate | ||||||
 要素 要素 | Isoaspartyl dipeptidase | ||||||
 キーワード キーワード | HYDROLASE / amidohydrolase / metalloprotease | ||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報加水分解酵素; プロテアーゼ; ペプチド結合加水分解酵素; オメガペプチターゼ / hydrolase activity, acting on carbon-nitrogen (but not peptide) bonds / beta-aspartyl-peptidase activity / metallopeptidase activity / proteolysis / zinc ion binding / identical protein binding / cytoplasm / cytosol 類似検索 - 分子機能 | ||||||
| 生物種 |  | ||||||
| 手法 |  X線回折 / 分子置換 / 解像度: 2.1 Å X線回折 / 分子置換 / 解像度: 2.1 Å | ||||||
 データ登録者 データ登録者 | Thoden, J.B. / Marti-Arbona, R. / Raushel, F.M. / Holden, H.M. | ||||||
 引用 引用 | ジャーナル: Biochemistry / 年: 2003 タイトル: High Resolution X-ray Structure of Isoaspartyl Dipeptidase from Escherichia coli 著者: Thoden, J.B. / Marti-Arbona, R. / Raushel, F.M. / Holden, H.M. | ||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 1onx.cif.gz | 161.3 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb1onx.ent.gz | 126.6 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 1onx.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/on/1onxftp://data.pdbj.org/pub/pdb/validation_reports/on/1onx | HTTPS FTP |
|---|
-関連構造データ











-リンク
 PDBj
PDBj


- 集合体
集合体
| 登録構造単位 | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 単位格子 |
| ||||||||
| 詳細 | The biological unit is an octamer. It is generated by expanding the crystallographic independent unit around the crystallographic 4-fold axis using the following three transformations: (I)TRANSLATION VECTOR IN fractions of cell edge 0.500 0.500 0.000 ROTATION MATRIX 0.000 1.000 0.000 -1.000 0.000 0.000 0.000 0.000 1.000 (II)TRANSLATION VECTOR IN fractions of cell edge 1.000 0.000 0.000 ROTATION MATRIX -1.000 0.000 0.000 0.000 -1.000 0.000 0.000 0.000 1.000 (III)TRANSLATION VECTOR IN fractions of cell edge 0.500 -0.500 0.000 ROTATION MATRIX 0.000 -1.000 0.000 1.000 0.000 0.000 0.000 0.000 1.000 |
-要素
| #1: タンパク質 | 分子量: 41167.758 Da / 分子数: 2 / 由来タイプ: 組換発現 / 由来: (組換発現) 参照: UniProt: P39377, 加水分解酵素; プロテアーゼ; ペプチド結合加水分解酵素; オメガペプチターゼ #2: 化合物 | ChemComp-ZN /   分子量: 65.409 Da / 分子数: 4 / 由来タイプ: 合成 / 式: Zn 分子量: 65.409 Da / 分子数: 4 / 由来タイプ: 合成 / 式: Zn#3: 化合物 | 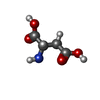  タイプ: L-peptide linking / 分子量: 133.103 Da / 分子数: 2 / 由来タイプ: 合成 / 式: C4H7NO4 タイプ: L-peptide linking / 分子量: 133.103 Da / 分子数: 2 / 由来タイプ: 合成 / 式: C4H7NO4#4: 水 | ChemComp-HOH / |  分子量: 18.015 Da / 分子数: 257 / 由来タイプ: 天然 / 式: H2O 分子量: 18.015 Da / 分子数: 257 / 由来タイプ: 天然 / 式: H2O |
|---|
-実験情報
-実験
| 実験 | 手法: X線回折 / 使用した結晶の数: 1 |
|---|
- 試料調製
試料調製
| 結晶 | マシュー密度: 2.89 Å3/Da / 溶媒含有率: 57.1 % | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 結晶化 | 温度: 295 K / 手法: 蒸気拡散法, ハンギングドロップ法 / pH: 5 詳細: PEG8000, homopipes, magnesium chloride, pH 5.0, VAPOR DIFFUSION, HANGING DROP, temperature 295K | ||||||||||||||||||||||||||||||||||||||||||
| 結晶化 | *PLUS 温度: 4 ℃ | ||||||||||||||||||||||||||||||||||||||||||
| 溶液の組成 | *PLUS
|
-データ収集
| 回折 | 平均測定温度: 277 K |
|---|---|
| 放射光源 | 由来: 回転陽極 / タイプ: RIGAKU RU200 / 波長: 1.5418 Å |
| 検出器 | タイプ: SIEMENS HI-STAR / 検出器: AREA DETECTOR / 日付: 2003年1月20日 / 詳細: supper "long" mirrors |
| 放射 | モノクロメーター: Ni FILTER / プロトコル: SINGLE WAVELENGTH / 単色(M)・ラウエ(L): M / 散乱光タイプ: x-ray |
| 放射波長 | 波長: 1.5418 Å / 相対比: 1 |
| 反射 | 解像度: 2.1→30 Å / Num. all: 55201 / Num. obs: 55201 / % possible obs: 94.5 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / 冗長度: 4.2 % / Rsym value: 0.082 / Net I/σ(I): 6.7 |
| 反射 シェル | 解像度: 2.1→2.2 Å / 冗長度: 1.9 % / Mean I/σ(I) obs: 1.6 / Num. unique all: 5640 / Rsym value: 0.263 / % possible all: 75.4 |
| 反射 | *PLUS Rmerge(I) obs: 0.082 |
| 反射 シェル | *PLUS % possible obs: 75.4 % / Num. unique obs: 5640 / Rmerge(I) obs: 0.263 |
- 解析
解析
| ソフトウェア |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 精密化 | 構造決定の手法: 分子置換 開始モデル: 1ONW 解像度: 2.1→30 Å / 交差検証法: THROUGHOUT / σ(F): 0 / 立体化学のターゲット値: Engh & Huber
| |||||||||||||||||||||||||
| 精密化ステップ | サイクル: LAST / 解像度: 2.1→30 Å
| |||||||||||||||||||||||||
| 拘束条件 |
| |||||||||||||||||||||||||
| ソフトウェア | *PLUS 名称: 'TNT V.' / 分類: refinement | |||||||||||||||||||||||||
| 精密化 | *PLUS 最高解像度: 2.1 Å / Num. reflection obs: 49618 / Rfactor all: 0.182 | |||||||||||||||||||||||||
| 溶媒の処理 | *PLUS | |||||||||||||||||||||||||
| 原子変位パラメータ | *PLUS | |||||||||||||||||||||||||
| 拘束条件 | *PLUS
|
