 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1ola: THE STRUCTURAL BASIS OF MULTISPECIFICITY IN THE OLIGOPEPTIDE-BIND... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1ola | ||||||
|---|---|---|---|---|---|---|---|
| Title | THE STRUCTURAL BASIS OF MULTISPECIFICITY IN THE OLIGOPEPTIDE-BINDING PROTEIN OPPA | ||||||
 Components Components |
| ||||||
 Keywords Keywords | BINDING PROTEIN | ||||||
| Function / homology |  Function and homology information Function and homology informationpeptide transport / peptide transmembrane transporter activity / ATP-binding cassette (ABC) transporter complex / protein transport / outer membrane-bounded periplasmic space Similarity search - Function | ||||||
| Biological species |  Salmonella typhimurium (bacteria) Salmonella typhimurium (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2.1 Å X-RAY DIFFRACTION / Resolution: 2.1 Å | ||||||
 Authors Authors | Tame, J. / Wilkinson, A.J. | ||||||
 Citation Citation | Journal: Science / Year: 1994 Title: The structural basis of sequence-independent peptide binding by OppA protein. Authors: Tame, J.R. / Murshudov, G.N. / Dodson, E.J. / Neil, T.K. / Dodson, G.G. / Higgins, C.F. / Wilkinson, A.J. #1: Journal: To be PublishedTitle: Structure Determination of Oppa at 2.3 Angstroms Resolution Using Multiple Wavelength Anomalous Methods Authors: Glover, I.D. / Denny, R. / Nguti, N.D. / Mcsweeney, S. / Thompson, A. / Dodson, E. / Wilkinson, A.J. / Tame, J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1ola.cif.gz | 123.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1ola.ent.gz | 95.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1ola.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ol/1olaftp://data.pdbj.org/pub/pdb/validation_reports/ol/1ola | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|













-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Atom site foot note | 1: CIS PROLINE - PRO A 283 |
-Components
| #1: Protein | Mass: 58878.984 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Salmonella typhimurium (bacteria) / References: UniProt: P06202 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| #2: Protein/peptide | Mass: 400.492 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source | ||||||||
| #3: Chemical | 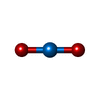  Mass: 270.028 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: O2U Mass: 270.028 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: O2U#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 354 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 354 / Source method: isolated from a natural source / Formula: H2OCompound details | THERE ARE NO DISCONTINU | Has protein modification | Y | Nonpolymer details | THE PROTEIN WAS CO-CRYSTALLIZED WITH URANYL (VI) ACETATE. TWO URANIUM ATOMS ARE INCLUDED IN THE ...THE PROTEIN WAS CO-CRYSTALLIZ | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.34 Å3/Da / Density % sol: 47.46 % | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS pH: 5.5 / Method: unknown | ||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Radiation | Scattering type: x-ray |
|---|---|
| Radiation wavelength | Relative weight: 1 |
| Reflection | *PLUS Highest resolution: 2.1 Å / Num. obs: 30656 / % possible obs: 89.7 % / Rmerge(I) obs: 0.067 |
- Processing
Processing
| Software | Name: PROLSQ / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.1→8 Å / σ(F): 0 Details: CRYSTALS OF OPPA ARE ONLY OBTAINED IN THE PRESENCE OF URANYL IONS WHICH BIND TO THE PROTEIN AND ARE RESPONSIBLE FOR FORMING IMPORTANT CRYSTAL CONTACTS. TWO CRYSTAL FORMS OF URANYL OPPA HAVE ...Details: CRYSTALS OF OPPA ARE ONLY OBTAINED IN THE PRESENCE OF URANYL IONS WHICH BIND TO THE PROTEIN AND ARE RESPONSIBLE FOR FORMING IMPORTANT CRYSTAL CONTACTS. TWO CRYSTAL FORMS OF URANYL OPPA HAVE BEEN FOUND. OPPA CONTAINING CO-PURIFIED PEPTIDES CO-CRYSTALLIZED WITH URANYL ACETATE GIVES CRYSTALS IN SPACE-GROUP P 21 21 2 CONTAINING TWO URANIUM ATOMS. ONE URANIUM ATOM FORMS AN IMPORTANT CRYSTAL CONTACT. THE OTHER LIES VERY CLOSE TO THE TWO-FOLD AXIS AND HAS A HIGH TEMPERATURE FACTOR. NO ATTEMPT WAS MADE IN REFINEMENT TO CONSTRAIN THIS ATOM TO THE SYMMETRY AXIS. IT HAS BEEN ASSIGNED AN OCCUPANCY OF 0.5 TO ALLOW FOR SYMMETRY. OPPA LIGANDED WITH TRI-LYSINE CRYSTALLIZES WITH EIGHT URANYL IONS PER PROTEIN MOLECULE IN SPACE-GROUP P 21 21 21. THIS STRUCTURE IS PRESENTED IN PDB ENTRY 1OLB.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.1→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: PROLSQ / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Num. reflection all: 29634 / Rfactor all: 0.167 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
