 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1dpp | ||||||
|---|---|---|---|---|---|---|---|
| Title | DIPEPTIDE BINDING PROTEIN COMPLEX WITH GLYCYL-L-LEUCINE | ||||||
 Components Components | DIPEPTIDE BINDING PROTEIN | ||||||
 Keywords Keywords | PEPTIDE BINDING PROTEIN / CHEMOTAXIS / COMPLEX (BINDING PROTEIN-PEPTIDE) COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationdipeptide transport / heme transport / heme transmembrane transport / dipeptide transmembrane transporter activity / peptide transmembrane transporter activity / Heme assimilation / positive chemotaxis / ATP-binding cassette (ABC) transporter complex, substrate-binding subunit-containing / peptide binding / protein transport ...dipeptide transport / heme transport / heme transmembrane transport / dipeptide transmembrane transporter activity / peptide transmembrane transporter activity / Heme assimilation / positive chemotaxis / ATP-binding cassette (ABC) transporter complex, substrate-binding subunit-containing / peptide binding / protein transport / outer membrane-bounded periplasmic space / heme binding / membrane Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 3.2 Å X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 3.2 Å | ||||||
 Authors Authors | Dunten, P. / Mowbray, S.L. | ||||||
 Citation Citation | Journal: Protein Sci. / Year: 1995 Title: Crystal structure of the dipeptide binding protein from Escherichia coli involved in active transport and chemotaxis. Authors: Dunten, P. / Mowbray, S.L. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1dpp.cif.gz | 357.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1dpp.ent.gz | 297.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1dpp.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/dp/1dppftp://data.pdbj.org/pub/pdb/validation_reports/dp/1dpp | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|
















-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||
| 2 | 
| ||||||||||||||||
| 3 | 
| ||||||||||||||||
| 4 | 
| ||||||||||||||||
| Unit cell |
| ||||||||||||||||
| Atom site foot note | 1: CIS PROLINE - PRO A 275 / 2: CIS PROLINE - PRO C 275 / 3: CIS PROLINE - PRO E 275 / 4: CIS PROLINE - PRO G 275 | ||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper:
|
-Components
| #1: Protein | Mass: 57475.863 Da / Num. of mol.: 4 / Source method: isolated from a natural source / Source: (natural) #2: Chemical | ChemComp-GLY / 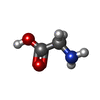  Type: peptide linking / Mass: 75.067 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H5NO2 Type: peptide linking / Mass: 75.067 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H5NO2#3: Chemical | ChemComp-LEU /   Type: L-peptide linking / Mass: 131.173 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C6H13NO2 Type: L-peptide linking / Mass: 131.173 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C6H13NO2Has protein modification | Y | Nonpolymer details | RESIDUES 1001 AND 1002 WITH CHAIN IDS A, C, E AND G FORM THE DIPEPTIDE SUBSTRATE BOUND TO THE ...RESIDUES 1001 AND 1002 WITH CHAIN IDS A, C, E AND G FORM THE DIPEPTIDE SUBSTRATE BOUND TO THE DIPEPTIDE-BINDING PROTEIN | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.42 Å3/Da / Density % sol: 72.15 % | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 6.2 / Details: pH 6.2 | |||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop | |||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction source | Source: SYNCHROTRON / Site: SRS  / Beamline: PX9.6 / Wavelength: 0.89 / Beamline: PX9.6 / Wavelength: 0.89 |
|---|---|
| Detector | Type: RIGAKU RAXIS II / Detector: IMAGE PLATE / Date: Feb 17, 1993 |
| Radiation | Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.89 Å / Relative weight: 1 |
| Reflection | Resolution: 3.2→25 Å / Num. obs: 57483 / % possible obs: 88 % / Redundancy: 3 % / Rmerge(I) obs: 0.105 |
| Reflection | *PLUS Num. measured all: 170663 / Rmerge(I) obs: 0.105 |
| Reflection shell | *PLUS Highest resolution: 3.2 Å / Lowest resolution: 3.28 Å / Mean I/σ(I) obs: 2.6 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 3.2→25 Å / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 21.5 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.2→25 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
