 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1b32 | ||||||
|---|---|---|---|---|---|---|---|
| Title | OLIGO-PEPTIDE BINDING PROTEIN (OPPA) COMPLEXED WITH KMK | ||||||
 Components Components |
| ||||||
 Keywords Keywords | PEPTIDE BINDING PROTEIN / COMPLEX (PEPTIDE TRANSPORT-PEPTIDE) / PEPTIDE TRANSPORT | ||||||
| Function / homology |  Function and homology information Function and homology informationpeptide transport / peptide transmembrane transporter activity / ATP-binding cassette (ABC) transporter complex / protein transport / outer membrane-bounded periplasmic space Similarity search - Function | ||||||
| Biological species |  Salmonella typhimurium (bacteria) Salmonella typhimurium (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / OTHER / Resolution: 1.75 Å X-RAY DIFFRACTION / SYNCHROTRON / OTHER / Resolution: 1.75 Å | ||||||
 Authors Authors | Tame, J.R.H. / Wilkinson, A.J. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 1999 Title: Crystallographic and calorimetric analysis of peptide binding to OppA protein. Authors: Sleigh, S.H. / Seavers, P.R. / Wilkinson, A.J. / Ladbury, J.E. / Tame, J.R. #1: Journal: Nat.Struct.Biol. / Year: 1996Title: The Role of Water in Sequence-Independent Ligand Binding by an Oligopeptide Transporter Protein Authors: Tame, J.R.H. / Sleigh, S.H. / Wilkinson, A.J. / Ladbury, J.E. #2: Journal: Structure / Year: 1995Title: The Crystal Structures of the Oligopeptide-Binding Protein OppA Complexed with Tripeptide and Tetrapeptide Ligands Authors: Tame, J.R.H. / Dodson, E.J. / Murshudov, G. / Higgins, C.F. / Wilkinson, A.J. #3: Journal: Acta Crystallogr.,Sect.D / Year: 1995Title: Structure Determination of Oppa at 2.3 Angstroms Resolution Using Multiple Wavelength Anomalous Methods Authors: Glover, I.D. / Denny, R. / Nguti, N.D. / McSweeney, S. / Thompson, A. / Dodson, E. / Wilkinson, A.J. / Tame, J.R.H. #4: Journal: Science / Year: 1994Title: The Structural Basis of Sequence-Independent Peptide Binding by OppA Protein Authors: Tame, J.R.H. / Murshudov, G.N. / Dodson, E.J. / Neil, T.K. / Dodson, G.G. / Higgins, C.F. / Wilkinson, A.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1b32.cif.gz | 127.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1b32.ent.gz | 97.6 KB | Display | PDB format |
| PDBx/mmJSON format | 1b32.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/b3/1b32ftp://data.pdbj.org/pub/pdb/validation_reports/b3/1b32 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1b05C  1b3fC  1b3gC  1b3lC  1b40C  1b46C  1b4zC  1b51C  1b52C  1b58C  1b5iC  1b5jC  1b9jC  1qkaC  1qkbC  1olb S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 58878.984 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Salmonella typhimurium (bacteria) / Gene: OPPA / Production host: | ||||||
|---|---|---|---|---|---|---|---|
| #2: Protein/peptide | Mass: 407.571 Da / Num. of mol.: 1 / Source method: obtained synthetically | ||||||
| #3: Chemical | ChemComp-IUM / 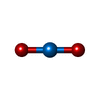  Mass: 270.028 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: O2U Mass: 270.028 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: O2U#4: Chemical | ChemComp-ACT /   Mass: 59.044 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C2H3O2 Mass: 59.044 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C2H3O2#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 437 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 437 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.46 Å3/Da / Density % sol: 45 % / Description: FLASH COOLED TO 120K | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 5.5 / Details: CO-CRYSTALLIZED WITH URANIUM ACETATE, PH 5.5 | |||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop | |||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 120 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: X11 / Wavelength: 0.97 / Beamline: X11 / Wavelength: 0.97 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97 Å / Relative weight: 1 |
| Reflection | Resolution: 1.75→44.2 Å / Num. obs: 60799 / % possible obs: 96.69 % / Observed criterion σ(I): 0 / Redundancy: 3.3 % / Biso Wilson estimate: 14.2 Å2 / Rmerge(I) obs: 0.059 |
| Reflection shell | Resolution: 1.75→1.78 Å / Rmerge(I) obs: 0.203 / Mean I/σ(I) obs: 4 / % possible all: 99.2 |
| Reflection shell | *PLUS % possible obs: 99.2 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: OTHER Starting model: PDB ENTRY 1OLB 1olb Resolution: 1.75→15 Å / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 17.7 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.75→15 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: REFMAC / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor Rwork: 0.185 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
