 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1obo | ||||||
|---|---|---|---|---|---|---|---|
| Title | W57L flavodoxin from Anabaena | ||||||
 Components Components | FLAVODOXIN | ||||||
 Keywords Keywords | ELECTRON TRANSFER / FLAVOPROTEIN / ELECTRON TRANSPORT | ||||||
| Function / homology |  Function and homology information Function and homology informationcellular response to iron ion starvation / electron transport chain / FMN binding / electron transfer activity Similarity search - Function | ||||||
| Biological species |  ANABAENA SP. (bacteria) ANABAENA SP. (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.2 Å | ||||||
 Authors Authors | Romero, A. / Ramon, A. / Fernandez-Cabrera, C. / Irun, M.P. / Sancho, J. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2003 Title: How Fmn Binds to Anabaena Apoflavodoxin: A Hydrophobic Encounter at an Open Binding Site Authors: Lostao, A. / Daoudi, F. / Irun, M.P. / Ramon, A. / Fernandez-Cabrera, C. / Romero, A. / Sancho, J. #1: Journal: Nat.Struct.Biol. / Year: 1996Title: Closure of a Tyrosine/Tryptophane Aromatic Gate Leeds to a Compact Fold in Apoflavodoxin Authors: Genzor, C.G. / Perales-Alcon, A. / Sancho, J. / Romero, A. | ||||||
| History |
| ||||||
| Remark 700 | SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN ... SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN THE SHEET RECORDS BELOW, TWO SHEETS ARE DEFINED. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1obo.cif.gz | 88 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1obo.ent.gz | 65.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1obo.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ob/1oboftp://data.pdbj.org/pub/pdb/validation_reports/ob/1obo | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1obvC  1flvS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |














-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (0.86226, 0.50568, 0.02814), Vector: |
-Components
| #1: Protein | Mass: 18758.477 Da / Num. of mol.: 2 / Mutation: YES Source method: isolated from a genetically manipulated source Source: (gene. exp.) ANABAENA SP. (bacteria) / Strain: PCC 7119 / Cell line: E.COLI JM109 / Plasmid: PTRC99A / Production host: #2: Chemical | 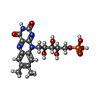  Mass: 456.344 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C17H21N4O9P Mass: 456.344 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C17H21N4O9P#3: Chemical | ChemComp-SO4 / |   Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 309 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 309 / Source method: isolated from a natural source / Formula: H2OCompound details | ENGINEERED MUTATION TRP 57 LEU THE PROTEIN IS A LOW-POTENTIAL ELECTRON DONOR TO A NUMBER OF ENZYMES. ...ENGINEERED | Sequence details | THE CONFLICT SHOWN IN THE SEQADV RECORDS BELOW ARISE FROM THE CLONING PROCESS AND HAS BEEN ...THE CONFLICT SHOWN IN THE SEQADV RECORDS BELOW ARISE FROM THE CLONING PROCESS AND HAS BEEN DESCRIBED IN M.F. FILLAT, W.E. BORRIAS, P.J. WEISBEEK, BIOCHEM. J. 280, 187-191 (1991) | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.03 Å3/Da / Density % sol: 39.03 % | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 6 / Details: pH 6.00 | ||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 295 K / Method: vapor diffusion, sitting drop | ||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: X11 / Wavelength: 0.9102 / Beamline: X11 / Wavelength: 0.9102 |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Nov 15, 2002 / Details: MIRRORS |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9102 Å / Relative weight: 1 |
| Reflection | Resolution: 1.2→30 Å / Num. obs: 88832 / % possible obs: 94.7 % / Redundancy: 3.6 % / Biso Wilson estimate: 7.2 Å2 / Rmerge(I) obs: 0.037 / Net I/σ(I): 10.8 |
| Reflection shell | Resolution: 1.2→1.26 Å / Redundancy: 3.1 % / Rmerge(I) obs: 0.138 / Mean I/σ(I) obs: 3.8 / % possible all: 92.1 |
| Reflection | *PLUS Lowest resolution: 30 Å / Num. obs: 81435 / Num. measured all: 200831 / Rmerge(I) obs: 0.038 |
| Reflection shell | *PLUS % possible obs: 92.1 % / Num. unique obs: 11496 / Num. measured obs: 28242 / Rmerge(I) obs: 0.139 / Mean I/σ(I) obs: 3.6 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1FLV Resolution: 1.2→19.86 Å / Rfactor Rfree error: 0.003 / Data cutoff high absF: 1008568.31 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 45.7723 Å2 / ksol: 0.402019 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 10.5 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.2→19.86 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.2→1.28 Å / Rfactor Rfree error: 0.007 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 20 Å / Rfactor Rfree: 0.203 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
