 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1o0c: CRYSTAL STRUCTURE OF L-GLUTAMATE AND AMPCPP BOUND TO GLUTAMINE AM... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1o0c | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF L-GLUTAMATE AND AMPCPP BOUND TO GLUTAMINE AMINOACYL TRNA SYNTHETASE | ||||||
 Components Components |
| ||||||
 Keywords Keywords | LIGASE/RNA / ENGINEERED TRNA / TRNA-PROTEIN COMPLEX / amino acid specificity / LIGASE-RNA COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationglutamine-tRNA ligase / glutamine-tRNA ligase activity / glutaminyl-tRNA aminoacylation / glutamyl-tRNA aminoacylation / ATP binding / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 2.7 Å X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 2.7 Å | ||||||
 Authors Authors | Bullock, T.L. / Perona, J.J. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2003 Title: Amino Acid Discrimination by a class I aminoacyl-tRNA synthetase specified by negative determinants Authors: Bullock, T.L. / Uter, N. / Nissan, T.A. / Perona, J.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1o0c.cif.gz | 165.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1o0c.ent.gz | 125.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1o0c.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/o0/1o0cftp://data.pdbj.org/pub/pdb/validation_reports/o0/1o0c | HTTPS FTP |
|---|
-Related structure data









-Links
 PDBj
PDBj



































- Assembly
Assembly
| Deposited unit | 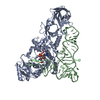
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||
| Unit cell |
|
-Components
-RNA chain / Protein , 2 types, 2 molecules BA
| #1: RNA chain | Mass: 24060.287 Da / Num. of mol.: 1 / Mutation: U1G to facilitate in vitro transcription / Source method: obtained synthetically Details: PRODUCT OF RUNOFF T7 POLYMERASE TRANSCRIPTION FROM A DOUBLE HELICAL DNA TEMPLATE References: EMBL: 43058 |
|---|---|
| #2: Protein | Mass: 63565.836 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Details: complexed with AMPCPP, see REMARK 600 / Source: (gene. exp.) References: UniProt: P00962, UniProt: A0A140NAB7*PLUS, glutamine-tRNA ligase |
-Non-polymers , 4 types, 148 molecules 
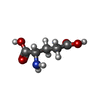


| #3: Chemical |  Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4#4: Chemical | ChemComp-GLU / |  Type: L-peptide linking / Mass: 147.129 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H9NO4 Type: L-peptide linking / Mass: 147.129 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H9NO4#5: Chemical | ChemComp-AMP / |  Mass: 347.221 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H14N5O7P / Comment: AMP*YM Mass: 347.221 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H14N5O7P / Comment: AMP*YM#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 144 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.77 Å3/Da / Density % sol: 67.41 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 7 Details: Ammonium Sulfate, pH 7.00, VAPOR DIFFUSION, HANGING DROP, temperature 298K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 7 / Method: vapor diffusion | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SRS  / Beamline: PX9.6 / Wavelength: 1 Å / Beamline: PX9.6 / Wavelength: 1 Å |
| Detector | Type: UCSD MARK II / Detector: AREA DETECTOR / Date: Jun 15, 2000 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.5→41 Å / Num. all: 46224 / Num. obs: 45855 / % possible obs: 99.2 % / Observed criterion σ(I): 2 / Redundancy: 3.5 % / Biso Wilson estimate: 55.4 Å2 / Rmerge(I) obs: 0.07 / Rsym value: 0.06 / Net I/σ(I): 8.9 |
| Reflection shell | Resolution: 2.5→2.8 Å / Rmerge(I) obs: 0.529 / % possible all: 99.3 |
| Reflection | *PLUS Highest resolution: 2.5 Å / Lowest resolution: 41.2 Å / Num. measured all: 159330 / Rmerge(I) obs: 0.07 |
| Reflection shell | *PLUS % possible obs: 99.2 % |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS / Resolution: 2.7→6 Å / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber
| |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.7→6 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.5 Å / Lowest resolution: 6 Å / % reflection Rfree: 5 % / Rfactor Rfree: 0.279 / Rfactor Rwork: 0.221 | |||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||
| Displacement parameters | *PLUS |
