 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1nd1: Amino acid sequence and crystal structure of BaP1, a metalloprote... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1nd1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Amino acid sequence and crystal structure of BaP1, a metalloproteinase from Bothrops asper snake venom that exerts multiple tissue-damaging activities. | |||||||||
 Components Components | BaP1 | |||||||||
 Keywords Keywords | TOXIN / metalloproteinase / snake venom / three-disulfide | |||||||||
| Function / homology |  Function and homology information Function and homology informationHydrolases; Acting on peptide bonds (peptidases); Metalloendopeptidases / metalloendopeptidase activity / chemotaxis / toxin activity / proteolysis / extracellular region / metal ion binding / plasma membrane Similarity search - Function | |||||||||
| Biological species |  Bothrops asper (terciopelo) Bothrops asper (terciopelo) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.93 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.93 Å | |||||||||
 Authors Authors | Watanabe, L. / Shannon, J.D. / Valente, R.H. / Rucavado, A. / Alape-Giron, A. / Kamiguti, A.S. / Theakston, R.D. / Fox, J.W. / Gutierrez, J.M. / Arni, R.K. | |||||||||
 Citation Citation | Journal: Protein Sci. / Year: 2003 Title: Amino acid sequence and crystal structure of BaP1, a metalloproteinase from Bothrops asper snake venom that exerts multiple tissue-damaging activities Authors: Watanabe, L. / Shannon, J.D. / Valente, R.H. / Rucavado, A. / Alape-Giron, A. / Kamiguti, A.S. / Theakston, R.D. / Fox, J.W. / Gutierrez, J.M. / Arni, R.K. #1: Journal: Acta Crystallogr.,Sect.D / Year: 1999Title: Structure of acutolysin-C, a haemorrhagic toxin from the venom of Agkistrodon acutus, providing further evidence for the mechanism of the pH-dependent proteolytic reaction of zinc metalloproteinases. Authors: Zhu, X. / Teng, M. / Niu, L. | |||||||||
| History |
|
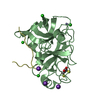
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1nd1.cif.gz | 55.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1nd1.ent.gz | 39.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1nd1.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/nd/1nd1ftp://data.pdbj.org/pub/pdb/validation_reports/nd/1nd1 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1quaS S: Starting model for refinement |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 22747.682 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Bothrops asper (terciopelo) / Secretion: Venom / References: UniProt: P83512 |
|---|---|
| #2: Chemical | ChemComp-ZN /   Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 110 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 110 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.09 Å3/Da / Density % sol: 40.77 % | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 9 Details: PEG 20000, dioxane, pH 9.0, VAPOR DIFFUSION, HANGING DROP, temperature 298K | ||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 298 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: LNLS  / Beamline: D03B-MX1 / Wavelength: 1.5418 Å / Beamline: D03B-MX1 / Wavelength: 1.5418 Å |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Jul 23, 2002 |
| Radiation | Monochromator: Graphite / Protocol: LAUE / Monochromatic (M) / Laue (L): L / Scattering type: neutron |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→500 Å / Num. all: 14941 / Num. obs: 49843 / % possible obs: 92.1 % / Biso Wilson estimate: 20 Å2 / Rsym value: 0.072 |
| Reflection shell | Resolution: 1.9→1.97 Å / Num. unique all: 816 / Rsym value: 0.28 / % possible all: 51.6 |
| Reflection | *PLUS Highest resolution: 1.9 Å / Num. obs: 14941 / Num. measured all: 49843 / Rmerge(I) obs: 0.072 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB_ID 1QUA Resolution: 1.93→18.31 Å / Rfactor Rfree error: 0.008 / Data cutoff high absF: 1182665.85 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0
| ||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 47.7738 Å2 / ksol: 0.306696 e/Å3 | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 29.7 Å2
| ||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.93→18.31 Å
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.9→2.02 Å / Rfactor Rfree error: 0.029 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.9 Å / % reflection Rfree: 10 % / Rfactor Rfree: 0.2159 / Rfactor Rwork: 0.1908 | ||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
