 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1n0s | ||||||
|---|---|---|---|---|---|---|---|
| Title | ENGINEERED LIPOCALIN FLUA IN COMPLEX WITH FLUORESCEIN | ||||||
 Components Components | Bilin-binding protein | ||||||
 Keywords Keywords | BINDING PROTEIN / PIERIS BRASSICAE / LIPOCALIN / ANTICALIN / PROTEIN ENGINEERING / fluorescein | ||||||
| Function / homology |  Function and homology information Function and homology informationpigment binding / response to reactive oxygen species / lipid metabolic process / extracellular region / cytoplasm Similarity search - Function | ||||||
| Biological species |  Pieris brassicae (large cabbage white) Pieris brassicae (large cabbage white) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2 Å | ||||||
 Authors Authors | Korndoerfer, I.P. / Skerra, A. | ||||||
 Citation Citation | Journal: Proteins / Year: 2003 Title: Crystallographic analysis of an "anticalin" with tailored specificity for fluorescein reveals high structural plasticity of the lipocalin loop region. Authors: Korndorfer, I.P. / Beste, G. / Skerra, A. | ||||||
| History |
| ||||||
| Remark 999 | SEQUENCE Swiss-Prot entry P09464 BBP_PIEBR is the sequence of the Bilin-binding protein (BBP) from ...SEQUENCE Swiss-Prot entry P09464 BBP_PIEBR is the sequence of the Bilin-binding protein (BBP) from P. brassicae, from wich this variant was created. Therefore, there are a number of residues which do not match the Swiss-Prot database sequence. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1n0s.cif.gz | 87 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1n0s.ent.gz | 65.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1n0s.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/n0/1n0sftp://data.pdbj.org/pub/pdb/validation_reports/n0/1n0s | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1kxoS S: Starting model for refinement |
|---|---|
| Similar structure data |













-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Ens-ID: 1 / Refine code: 6
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | FluA is a monomer in solution. The crystallographic dimer has probably no physiological relevance. |
-Components
| #1: Protein | Mass: 21059.465 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Pieris brassicae (large cabbage white) / Plasmid: pbbp21-flua / Production host:  #2: Chemical | 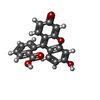  Mass: 332.306 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C20H12O5 Mass: 332.306 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C20H12O5#3: Chemical | ChemComp-SO4 / |   Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 98 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 98 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.3 Å3/Da / Density % sol: 46 % | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 8.1 Details: 2.0 M (NH4)2SO4, 2% (w/v) PEG 400, 10 mM hepes/naoh, pH 8.1, VAPOR DIFFUSION, HANGING DROP, temperature 298 KK | ||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU300 / Wavelength: 1.5418 Å |
|---|---|
| Detector | Type: MARRESEARCH / Detector: AREA DETECTOR / Date: May 15, 2001 / Details: osmic confocal maxflux |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2→99 Å / Num. all: 29552 / Num. obs: 29552 / % possible obs: 95.8 % / Redundancy: 5.39 % / Rsym value: 0.05 / Net I/σ(I): 17.64 |
| Reflection shell | Resolution: 2→2.07 Å / Redundancy: 2.73 % / Mean I/σ(I) obs: 2.73 / Num. unique all: 2956 / Rsym value: 0.47 / % possible all: 96.1 |
| Reflection | *PLUS Num. measured all: 79526 / Rmerge(I) obs: 0.05 |
| Reflection shell | *PLUS % possible obs: 96.1 % / Rmerge(I) obs: 0.47 / Mean I/σ(I) obs: 2.7 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1kxo Resolution: 2→63.25 Å / Cor.coef. Fo:Fc: 0.956 / Cor.coef. Fo:Fc free: 0.929 / SU B: 4.365 / SU ML: 0.121 / Cross valid method: THROUGHOUT / ESU R: 0.17 / ESU R Free: 0.163 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 34.125 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→63.25 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Dom-ID: 1 / Auth asym-ID: A / Ens-ID: 1 / Number: 1009 / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2→2.052 Å / Total num. of bins used: 20 /
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2 Å / Lowest resolution: 99 Å / Rfactor Rfree: 0.243 / Rfactor Rwork: 0.193 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
