 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1mxt: Atomic resolution structure of Cholesterol oxidase (Streptomyces ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1mxt | ||||||
|---|---|---|---|---|---|---|---|
| Title | Atomic resolution structure of Cholesterol oxidase (Streptomyces sp. SA-COO) | ||||||
 Components Components | CHOLESTEROL OXIDASE | ||||||
 Keywords Keywords | OXIDOREDUCTASE / FLAVOENZYME / STEROID METABOLISM / ATOMIC RESOLUTION | ||||||
| Function / homology |  Function and homology information Function and homology informationcholesterol oxidase / cholesterol oxidase activity / steroid Delta-isomerase / steroid Delta-isomerase activity / cholesterol catabolic process / flavin adenine dinucleotide binding / extracellular region Similarity search - Function | ||||||
| Biological species |  Streptomyces sp. (bacteria) Streptomyces sp. (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / AB INITIO PHASING / Resolution: 0.95 Å X-RAY DIFFRACTION / SYNCHROTRON / AB INITIO PHASING / Resolution: 0.95 Å | ||||||
 Authors Authors | Vrielink, A. / Lario, P.I. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2003 Title: Sub-atomic resolution crystal structure of cholesterol oxidase: What atomic resolution crystallography reveals about enzyme mechanism and the role of FAD cofactor in redox activity Authors: Lario, P.I. / Sampson, N. / Vrielink, A. #1: Journal: Nat.Chem.Biol. / Year: 2006Title: Atomic resolution crystallography reveals how changes in pH shape the protein microenvironment Authors: Lyubimov, A.Y. / Lario, P.I. / Moustafa, I. / Vrielink, A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1mxt.cif.gz | 342.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1mxt.ent.gz | 279.6 KB | Display | PDB format |
| PDBx/mmJSON format | 1mxt.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/mx/1mxtftp://data.pdbj.org/pub/pdb/validation_reports/mx/1mxt | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1b4vS S: Starting model for refinement |
|---|---|
| Similar structure data |














-Links
 PDBj
PDBj








- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 54969.676 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Details: FAD COFACTOR NON-COVALENTLY BOUND TO THE ENZYME / Source: (gene. exp.) Streptomyces sp. (bacteria) / Gene: CHOA / Plasmid: PCO202 / Production host: |
|---|---|
| #2: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 |
| #3: Chemical | ChemComp-FAE / 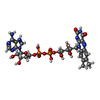  Mass: 786.558 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C27H34N9O15P2 Mass: 786.558 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C27H34N9O15P2 |
| #4: Chemical | ChemComp-OXY /   Mass: 31.999 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: O2 Mass: 31.999 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: O2 |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 738 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 738 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.7 Å3/Da / Density % sol: 40.39 % | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 290 K / Method: vapor diffusion, hanging drop / pH: 5 Details: PEG 8000, manganese sulfate, cacodylate, pH 5.0, VAPOR DIFFUSION, HANGING DROP, temperature 290K | ||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 17 ℃ / pH: 7 / Details: Yue, Q.K., (1999) Biochemistry, 38, 4277. | ||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X8C / Wavelength: 0.979 / Wavelength: 0.979 Å / Beamline: X8C / Wavelength: 0.979 / Wavelength: 0.979 Å |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Apr 23, 1999 |
| Radiation | Monochromator: CRYSTALS SI(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.979 Å / Relative weight: 1 |
| Reflection | Resolution: 0.95→28 Å / Num. obs: 266037 / % possible obs: 94.3 % / Redundancy: 3.9 % / Rmerge(I) obs: 0.058 / Net I/σ(I): 11.1 |
| Reflection shell | Resolution: 0.95→0.97 Å / Redundancy: 3.2 % / Rmerge(I) obs: 0.56 / Mean I/σ(I) obs: 1.5 / % possible all: 89.3 |
| Reflection | *PLUS Highest resolution: 0.95 Å / Lowest resolution: 28.2 Å / % possible obs: 94.1 % / Num. measured all: 1089496 / Rmerge(I) obs: 0.051 |
| Reflection shell | *PLUS % possible obs: 88 % |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: AB INITIO PHASING Starting model: 1B4V Resolution: 0.95→28 Å / Num. parameters: 45239 / Num. restraintsaints: 59298 / Cross valid method: FREE R / σ(F): 0 / Stereochemistry target values: ENGH AND HUBER Details: THE FOLLOWING ARE RESIDUES THAT WERE NOT LOCATED IN THE EXPERIMENT: ASP A 6, ASN A 7, GLY A 8, THR A 507 (only N was located), ALA A 508, SER A 509. SOME VERY MOBILE SIDE CHAINS WERE MODELED ...Details: THE FOLLOWING ARE RESIDUES THAT WERE NOT LOCATED IN THE EXPERIMENT: ASP A 6, ASN A 7, GLY A 8, THR A 507 (only N was located), ALA A 508, SER A 509. SOME VERY MOBILE SIDE CHAINS WERE MODELED AT EITHER 50% OR were not modelled. THE following atoms were not modelled: RES 146 NE->END is missing.; RES 183 CE->END is missing.; RES 436 CG->END is missing; PARTIALLY OCCUPIED SIDE CHAIN ATOMS MODELED AT 50% FOR THE FOLLOWING: RES 73 ATOMS CD OE1 OE2; RES 87 ATOMS CD 1HD 2HD NE HE CZ NH1 1HH1 2HH1 NH2 1HH2 2HH2; RES 127 ATOMS CG 1HG 2HG CE 1HE 2HE NZ 1HZ 2HZ 3HZ; RES 156 ATOMS CD 1HD 2HD NE HE CZ NH1 1HH1 2HH1 NH2 1HH2 2HH2; RES 163 ATOMS CG 1HG 2HG CD 1HD 2HD CE 1HE 2HE NZ 1HZ 2HZ 3HZ; RES 183 ATOMS CG 1HG 2HG CD 1HD 2HD; RES 241 ATOMS CE 1HE 2HE NZ 1HZ 2HZ 3HZ; RES 273 ATOMS CG 1HG 2HG CD 1HD 2HD CE 1HE 2HE NZ 1HZ 2HZ 3HZ; RES 365 ATOMS CG 1HG 2HG SD CE 1HE 2HE 3HE; RES 396 ATOMS CZ NH1 1HH1 2HH1 NH2 1HH2 2HH2; RES 398 ATOMS CE 1HE 2HE NZ 1HZ 2HZ 3HZ; RES 435 ATOMS CB HB OG1 CG2 1HG2 2HG2 3HG2; RES 436 ATOMS CB 1HB 2HB 3HB C; RES 468 ATOMS NZ 1HZ 2HZ 3HZ;
| |||||||||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 82 / Occupancy sum hydrogen: 3697.86 / Occupancy sum non hydrogen: 4440.8 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 0.95→28 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||
| Software | *PLUS Name: SHELXL / Version: 97 / Classification: refinement | |||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 28.2 Å / Num. reflection obs: 250371 / Rfactor Rfree: 0.132 / Rfactor Rwork: 0.11 | |||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
