 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1mjq: METHIONINE REPRESSOR MUTANT (Q44K) PLUS COREPRESSOR (S-ADENOSYL M... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1mjq | ||||||
|---|---|---|---|---|---|---|---|
| Title | METHIONINE REPRESSOR MUTANT (Q44K) PLUS COREPRESSOR (S-ADENOSYL METHIONINE) COMPLEXED TO AN ALTERED MET CONSENSUS OPERATOR SEQUENCE | ||||||
 Components Components |
| ||||||
 Keywords Keywords | TRANSCRIPTION/DNA / TRANSCRIPTION REGULATION / METJ / METHIONINE REPRESSOR / SHEET-HELIX-HELIX / S-ADENOSYL METHIONINE / DNA / COMPLEX (TRANSCRIPTION REGULATION-DNA) / TRANSCRIPTION-DNA COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology information: / DNA-binding transcription factor activity / negative regulation of DNA-templated transcription / DNA-templated transcription / DNA binding / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / DIFFERENCE FOURIER METHODS / Resolution: 2.4 Å X-RAY DIFFRACTION / SYNCHROTRON / DIFFERENCE FOURIER METHODS / Resolution: 2.4 Å | ||||||
 Authors Authors | Garvie, C.W. / Phillips, S.E.V. | ||||||
 Citation Citation | Journal: Structure Fold.Des. / Year: 2000 Title: Direct and indirect readout in mutant Met repressor-operator complexes. Authors: Garvie, C.W. / Phillips, S.E. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1mjq.cif.gz | 218.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1mjq.ent.gz | 178.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1mjq.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/mj/1mjqftp://data.pdbj.org/pub/pdb/validation_reports/mj/1mjq | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1mj2C  1mjmC  1mjoC  1mjpC  1cmaS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj






































- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: DNA chain | Mass: 5817.808 Da / Num. of mol.: 4 / Source method: obtained synthetically Details: SELF-COMPLIMENTARY SEQUENCE BASED ON THE UNDERLYING CONSENSUS SEQUENCE OF NATURALLY OCCURRING MET OPERATORS BUT WITH TWO CG BASE-STEPS MUTATED TO TA #2: Protein | Mass: 12028.607 Da / Num. of mol.: 8 / Mutation: Q44K Source method: isolated from a genetically manipulated source Source: (gene. exp.) #3: Chemical | ChemComp-SAM / 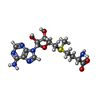  Mass: 398.437 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C15H22N6O5S Mass: 398.437 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C15H22N6O5S#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 186 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 186 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 2 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.91 Å3/Da / Density % sol: 58 % Description: THE FULL COMPLEX WAS USED AS A SEARCH MODEL FOR MOLECULAR REPLACEMENT, THE SECOND HALF OF THE METJ COMPLEX FROM 1CMA BEING GENERATED BY THE RELEVANT CRYSTALLOGRAPHIC TWO-FOLD. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7 Details: PROTEIN (10MG/ML) + SAM(1.5MG/ML) + DNA (4MG/ML) WAS CRYSTALLISED FROM 28-38% MPD, 100MM SODIUM CACODYLATE BUFFER, PH 6.0-7.0, 40MM CACL2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 7 / Method: vapor diffusion, sitting drop | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID2 / Wavelength: 0.99 / Beamline: ID2 / Wavelength: 0.99 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Apr 1, 1997 / Details: RHODIUM COATED MIRROR |
| Radiation | Monochromator: SILICON (111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.99 Å / Relative weight: 1 |
| Reflection | Resolution: 2.4→27.17 Å / Num. obs: 125697 / % possible obs: 97.8 % / Observed criterion σ(I): 0 / Redundancy: 2.4 % / Biso Wilson estimate: 40.8 Å2 / Rmerge(I) obs: 0.046 / Net I/σ(I): 10.4 |
| Reflection shell | Resolution: 2.4→2.53 Å / Redundancy: 1.9 % / Rmerge(I) obs: 0.046 / Mean I/σ(I) obs: 3.7 / Rsym value: 0.197 / % possible all: 97.8 |
| Reflection | *PLUS Num. obs: 51762 / Num. measured all: 125697 |
| Reflection shell | *PLUS % possible obs: 97.8 % / Rmerge(I) obs: 0.197 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: DIFFERENCE FOURIER METHODS Starting model: PDB ENTRY 1CMA Resolution: 2.4→27.3 Å / Rfactor Rfree error: 0.005 / Data cutoff high absF: 10000000 / Data cutoff low absF: 0.001 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 Details: BULK SOLVENT MODEL USED. IN GENERAL, ALL EIGHT MONOMERS DISPLAY LITTLE OR NO DENSITY FOR THE FIRST TWO RESIDUES AND A VARIED DEGREE OF DISCONTINUOUS AND/OR ILL-DEFINED DENSITY FOR THE ...Details: BULK SOLVENT MODEL USED. IN GENERAL, ALL EIGHT MONOMERS DISPLAY LITTLE OR NO DENSITY FOR THE FIRST TWO RESIDUES AND A VARIED DEGREE OF DISCONTINUOUS AND/OR ILL-DEFINED DENSITY FOR THE REGIONS 13 - 18, 76 - 83, AND 90 - 104.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 62.4 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.4→27.3 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.4→2.46 Å / Rfactor Rfree error: 0.045 / Total num. of bins used: 15
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.86 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
