 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1mcz: BENZOYLFORMATE DECARBOXYLASE FROM PSEUDOMONAS PUTIDA COMPLEXED WI... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1mcz | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | BENZOYLFORMATE DECARBOXYLASE FROM PSEUDOMONAS PUTIDA COMPLEXED WITH AN INHIBITOR, R-MANDELATE | |||||||||
 Components Components | BENZOYLFORMATE DECARBOXYLASE | |||||||||
 Keywords Keywords | LYASE / DECARBOXYLASE / THIAMIN DIPHOSPHATE / R-MANDELATE | |||||||||
| Function / homology |  Function and homology information Function and homology informationbenzoylformate decarboxylase / benzoylformate decarboxylase activity / (R)-mandelate catabolic process / acetolactate synthase activity / thiamine pyrophosphate binding / flavin adenine dinucleotide binding / magnesium ion binding Similarity search - Function | |||||||||
| Biological species |  Pseudomonas putida (bacteria) Pseudomonas putida (bacteria) | |||||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.8 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.8 Å | |||||||||
 Authors Authors | Polovnikova, E.S. / Bera, A.K. / Hasson, M.S. | |||||||||
 Citation Citation | Journal: Biochemistry / Year: 2003 Title: Structural and Kinetic Analysis of Catalysis by a Thiamin Diphosphate-Dependent Enzyme, Benzoylformate Decarboxylase Authors: POLOVNIKOVA, E.S. / McLeish, M.J. / Sergienko, E.A. / Burgner, J.T. / Anderson, N.L. / BERA, A.K. / Jordan, F. / Kenyon, G.L. / HASSON, M.S. #1: Journal: Biochemistry / Year: 1998Title: The crystal structure of benzoylformate decarboxylase at 1.6 A resolution: diversity of catalytic residues in thiamin diphosphate-dependent enzymes Authors: Hasson, M.S. / Muscate, A. / McLeish, M.J. / Polovnikova, L.S. / Gerlt, J.A. / Kenyon, G.L. / Petsko, G.A. / Ringe, D. | |||||||||
| History |
| |||||||||
| Remark 600 | HETEROGEN MG 529 (MAGNESIUM 2+) HAVE LOW B-FACTORS, SUGGESTING THAT THEY MAY ACTUALLY BE CALCIUM ...HETEROGEN MG 529 (MAGNESIUM 2+) HAVE LOW B-FACTORS, SUGGESTING THAT THEY MAY ACTUALLY BE CALCIUM IONS, AS IN STRUCTURE 1BFD. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1mcz.cif.gz | 1.5 MB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1mcz.ent.gz | 1.2 MB | Display | PDB format |
| PDBx/mmJSON format | 1mcz.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/mc/1mczftp://data.pdbj.org/pub/pdb/validation_reports/mc/1mcz | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1bfdS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| 4 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 56402.746 Da / Num. of mol.: 16 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Pseudomonas putida (bacteria) / Gene: MDLC / Plasmid: PKK233-2 / Production host: #2: Chemical | ChemComp-MG /   Mass: 24.305 Da / Num. of mol.: 24 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 24 / Source method: obtained synthetically / Formula: Mg#3: Chemical | ChemComp-TPP / 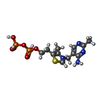  Mass: 425.314 Da / Num. of mol.: 16 / Source method: obtained synthetically / Formula: C12H19N4O7P2S Mass: 425.314 Da / Num. of mol.: 16 / Source method: obtained synthetically / Formula: C12H19N4O7P2S#4: Chemical | ChemComp-RMN / ( 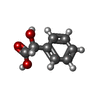  Mass: 152.147 Da / Num. of mol.: 16 / Source method: obtained synthetically / Formula: C8H8O3 Mass: 152.147 Da / Num. of mol.: 16 / Source method: obtained synthetically / Formula: C8H8O3#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 1656 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 1656 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.54 Å3/Da / Density % sol: 51.51 % | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 5.5 Details: CRYSTALS WERE GROWN AT ROOM TEMPERATURE BY HANGING-DROP VAPOR DIFFUSION AGAINST A WELL SOLUTION OF 20-22% PEG MME 2000, 100 mM Na citrate, pH 5.2-5.6, 0.15-0.2 M (NH4)2SO4 and 10 mM R- ...Details: CRYSTALS WERE GROWN AT ROOM TEMPERATURE BY HANGING-DROP VAPOR DIFFUSION AGAINST A WELL SOLUTION OF 20-22% PEG MME 2000, 100 mM Na citrate, pH 5.2-5.6, 0.15-0.2 M (NH4)2SO4 and 10 mM R-mandelate. DROPS CONTAINED EQUAL VOLUMES (2-4 MICROL) OF WELL SOLUTION AND PURIFIED BENZOYLFORMATE DECARBOXYLASE [20-50 MG/ML IN 0.1 MM MGCL2, 0.2 MM TDP, 15 MM NAHEPES (PH 7.0)]., pH 5.5, VAPOR DIFFUSION, HANGING DROP, temperature 293K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 20 ℃ / pH: 7 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 Å |
| Detector | Type: RIGAKU RAXIS IV / Detector: IMAGE PLATE |
| Radiation | Monochromator: CuKa / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.8→30 Å / Num. obs: 561773 / % possible obs: 93.6 % / Observed criterion σ(I): 2 / Rmerge(I) obs: 0.073 / Net I/σ(I): 9.1 |
| Reflection shell | Resolution: 2.8→2.9 Å / % possible all: 76.2 |
| Reflection | *PLUS Num. obs: 206940 / % possible obs: 94 % / Num. measured all: 561773 |
| Reflection shell | *PLUS % possible obs: 76 % / Rmerge(I) obs: 0.187 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1BFD Resolution: 2.8→30 Å / Isotropic thermal model: ANISOTROPIC / Cross valid method: THROUGHOUT / σ(F): 2 / Stereochemistry target values: Engh & Huber Details: NCS restraints were used on all 16 monomers from residues 2-460 and 472-525. From 461-471, NCS restraints were also applied to two groups: (1) monomers A, D, F, G, H, K and N and (2) ...Details: NCS restraints were used on all 16 monomers from residues 2-460 and 472-525. From 461-471, NCS restraints were also applied to two groups: (1) monomers A, D, F, G, H, K and N and (2) monomers B, C, E, H, I, L, M, O and P. The side chains of two residues, Phe 464 and Trp 463, are in different conformations in the two groups. In 7 out of 8 cases, the two monomers that compose two active sites in the tetramer have opposite conformations from each other.
| |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.8→30 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.8→2.82 Å
| |||||||||||||||||||||||||
| Refinement | *PLUS Rfactor Rwork: 0.2 | |||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
