- PDB-1m9p: Crystalline Human Carbonmonoxy Hemoglobin C Exhibits The R2 Quate... -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 1m9p
Title
Crystalline Human Carbonmonoxy Hemoglobin C Exhibits The R2 Quaternary State at Neutral pH In The Presence of Polyethylene Glycol: The 2.1 Angstrom Resolution Crystal Structure
Components
Hemoglobin alpha chain
Hemoglobin beta chain
Keywords
OXYGEN STORAGE/TRANSPORT / Mutant human hemoglobin C(beta E6K) / R2 quaternary state of human hemoglobin / OXYGEN STORAGE-TRANSPORT COMPLEX
Function / homology
Function and homology information
cellular oxidant detoxification / Heme assimilation / nitric oxide transport / hemoglobin alpha binding / hemoglobin binding / haptoglobin-hemoglobin complex / renal absorption / hemoglobin complex / oxygen transport / Scavenging of heme from plasma ...cellular oxidant detoxification / Heme assimilation / nitric oxide transport / hemoglobin alpha binding / hemoglobin binding / haptoglobin-hemoglobin complex / renal absorption / hemoglobin complex / oxygen transport / Scavenging of heme from plasma / erythrocyte development / endocytic vesicle lumen / blood vessel diameter maintenance / hydrogen peroxide catabolic process / oxygen carrier activity / carbon dioxide transport / response to hydrogen peroxide / Heme signaling / Erythrocytes take up oxygen and release carbon dioxide / Erythrocytes take up carbon dioxide and release oxygen / Cytoprotection by HMOX1 / oxygen binding / Late endosomal microautophagy / platelet aggregation / regulation of blood pressure / Chaperone Mediated Autophagy / positive regulation of nitric oxide biosynthetic process / tertiary granule lumen / Factors involved in megakaryocyte development and platelet production / blood microparticle / ficolin-1-rich granule lumen / iron ion binding / inflammatory response / heme binding / Neutrophil degranulation / : / extracellular exosome / extracellular region / membrane / metal ion binding / cytosol Similarity search - Function
COMPOUND THE QUATERNARY STRUCTURE OF THIS PDB ENTRY SUPERIMPOSES UPON THE R2 QUATERNARY STATE OF ...COMPOUND THE QUATERNARY STRUCTURE OF THIS PDB ENTRY SUPERIMPOSES UPON THE R2 QUATERNARY STATE OF COHBA (PDB ID 1BBB), BUT NOT UPON THE R QUATERNARY CONFORMATION OF OUR COHBC STRUCTURE OBTAINED IN CONCENTRATED PHOSPHATE BUFFER AT PH 7.35 (PDB ID 1K1K) OR UPON R-STATE COHBA(PDB ID 1HHO).
Remark 999
SEQUENCE Author states the protein was not genetically manipulated, but the residue E6K of chains B ...SEQUENCE Author states the protein was not genetically manipulated, but the residue E6K of chains B and D are allelic variants of human hemoglobin A.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads






















 PDBj
PDBj
















 Assembly
Assembly

 Mass: 616.487 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C34H32FeN4O4
Mass: 616.487 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C34H32FeN4O4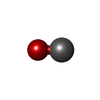
 Mass: 28.010 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: CO
Mass: 28.010 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: CO Mass: 18.015 Da / Num. of mol.: 437 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 437 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing