 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1kyd: AP-2 CLATHRIN ADAPTOR ALPHA-APPENDAGE IN COMPLEX WITH EPSIN DPW P... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1kyd | ||||||
|---|---|---|---|---|---|---|---|
| Title | AP-2 CLATHRIN ADAPTOR ALPHA-APPENDAGE IN COMPLEX WITH EPSIN DPW PEPTIDE | ||||||
 Components Components |
| ||||||
 Keywords Keywords | ENDOCYTOSIS/EXOCYTOSIS / PROTEIN-PEPTIDE COMPLEX / ENDOCYTOSIS / ENDOCYTOSIS-EXOCYTOSIS COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationnegative regulation of sprouting angiogenesis / clathrin vesicle coat / LDL clearance / WNT5A-dependent internalization of FZD2, FZD5 and ROR2 / WNT5A-dependent internalization of FZD4 / Trafficking of GluR2-containing AMPA receptors / Retrograde neurotrophin signalling / VLDLR internalisation and degradation / Recycling pathway of L1 / AP-2 adaptor complex ...negative regulation of sprouting angiogenesis / clathrin vesicle coat / LDL clearance / WNT5A-dependent internalization of FZD2, FZD5 and ROR2 / WNT5A-dependent internalization of FZD4 / Trafficking of GluR2-containing AMPA receptors / Retrograde neurotrophin signalling / VLDLR internalisation and degradation / Recycling pathway of L1 / AP-2 adaptor complex / postsynaptic neurotransmitter receptor internalization / Cargo recognition for clathrin-mediated endocytosis / membrane coat / Clathrin-mediated endocytosis / clathrin adaptor activity / clathrin-dependent endocytosis / MHC class II antigen presentation / regulation of hematopoietic stem cell differentiation / clathrin binding / molecular sequestering activity / synaptic vesicle endocytosis / embryonic organ development / vesicle-mediated transport / Notch signaling pathway / clathrin-coated pit / Neutrophil degranulation / secretory granule / intracellular protein transport / EGFR downregulation / female pregnancy / phospholipid binding / kinase binding / cytoplasmic side of plasma membrane / endocytosis / disordered domain specific binding / Cargo recognition for clathrin-mediated endocytosis / presynapse / Clathrin-mediated endocytosis / cytoplasmic vesicle / in utero embryonic development / postsynapse / endosome / protein domain specific binding / lipid binding / protein kinase binding / nucleus / plasma membrane / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2 Å | ||||||
 Authors Authors | Brett, T.J. / Traub, L.M. / Fremont, D.H. | ||||||
 Citation Citation | Journal: Structure / Year: 2002 Title: Accessory protein recruitment motifs in clathrin-mediated endocytosis. Authors: Brett, T.J. / Traub, L.M. / Fremont, D.H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1kyd.cif.gz | 66.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1kyd.ent.gz | 48 KB | Display | PDB format |
| PDBx/mmJSON format | 1kyd.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 1kyd_validation.pdf.gz | 381.1 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 1kyd_full_validation.pdf.gz | 383.3 KB | Display | |
| Data in XML | 1kyd_validation.xml.gz | 6.7 KB | Display | |
| Data in CIF | 1kyd_validation.cif.gz | 10.3 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ky/1kydftp://data.pdbj.org/pub/pdb/validation_reports/ky/1kyd | HTTPS FTP |
-Related structure data
| Related structure data |  1ky6C  1ky7C  1kyfC  1kyuC  1qtsS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |

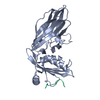
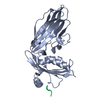
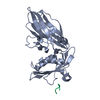







-Links
 PDBj
PDBj












- Assembly
Assembly
| Deposited unit | 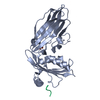
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 27828.676 Da / Num. of mol.: 1 / Fragment: C-TERMINAL APPENDAGE (EAR), RESIDUES 701-938 Source method: isolated from a genetically manipulated source Source: (gene. exp.)  |
|---|---|
| #2: Protein/peptide | Mass: 689.737 Da / Num. of mol.: 1 / Fragment: RESIDUES 341-345 / Mutation: T345K / Source method: obtained synthetically Details: The peptide was chemically synthesized. The sequence used (SPDWK) is a single mutation to the naturally occuring sequence from Human Epsin (SPDWT) References: GenBank: 5051636, UniProt: Q9Y6I3*PLUS |
| #3: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 180 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 180 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.11 Å3/Da / Density % sol: 41.66 % | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS pH: 6.8 / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 110 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU300 / Wavelength: 1.5418 Å |
| Detector | Type: RIGAKU RAXIS IV / Detector: IMAGE PLATE / Date: Nov 26, 2000 / Details: YALE MIRRORS |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2→50 Å / Num. all: 16055 / Num. obs: 16055 / % possible obs: 93.3 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Biso Wilson estimate: 22.3 Å2 / Rsym value: 0.0076 / Net I/σ(I): 12.4 |
| Reflection shell | Resolution: 2→2.07 Å / Mean I/σ(I) obs: 3.7 / Rsym value: 0.285 / % possible all: 84.1 |
| Reflection | *PLUS Lowest resolution: 50 Å / Num. measured all: 59931 / Rmerge(I) obs: 0.076 |
| Reflection shell | *PLUS % possible obs: 84.1 % / Rmerge(I) obs: 0.285 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1QTS Resolution: 2→19.73 Å / Rfactor Rfree error: 0.009 / Data cutoff high absF: 1530022.23 / Data cutoff high rms absF: 1530022.23 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): -3 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 61.8004 Å2 / ksol: 0.4041 e/Å3 | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 22.8 Å2
| ||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→19.73 Å
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2→2.13 Å / Rfactor Rfree error: 0.027 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.178 / Rfactor Rfree: 0.229 / Rfactor Rwork: 0.178 | ||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.271 / Rfactor Rwork: 0.201 / Rfactor obs: 0.201 |
