 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1kno: CRYSTAL STRUCTURE OF THE COMPLEX OF A CATALYTIC ANTIBODY FAB WITH... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1kno | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF THE COMPLEX OF A CATALYTIC ANTIBODY FAB WITH A TRANSITION STATE ANALOG: STRUCTURAL SIMILARITIES IN ESTERASE-LIKE ABZYMES | ||||||
 Components Components | (IGG2A FAB FRAGMENT CNJ206) x 2 | ||||||
 Keywords Keywords | CATALYTIC ANTIBODY | ||||||
| Function / homology |  Function and homology information Function and homology informationFc-gamma receptor I complex binding / alpha-beta T cell receptor complex / immunoglobulin complex, circulating / immunoglobulin receptor binding / IgG immunoglobulin complex / complement activation, classical pathway / antigen binding / B cell differentiation / antibacterial humoral response / adaptive immune response ...Fc-gamma receptor I complex binding / alpha-beta T cell receptor complex / immunoglobulin complex, circulating / immunoglobulin receptor binding / IgG immunoglobulin complex / complement activation, classical pathway / antigen binding / B cell differentiation / antibacterial humoral response / adaptive immune response / : / extracellular region / plasma membrane Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 3.2 Å X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 3.2 Å | ||||||
 Authors Authors | Charbonnier, J.-B. / Gigant, B. / Knossow, M. | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 1995 Title: Crystal structure of the complex of a catalytic antibody Fab fragment with a transition state analog: structural similarities in esterase-like catalytic antibodies. Authors: Charbonnier, J.B. / Carpenter, E. / Gigant, B. / Golinelli-Pimpaneau, B. / Eshhar, Z. / Green, B.S. / Knossow, M. #1: Journal: Mol.Immunol. / Year: 1994Title: Differences in the Biochemical Properties of Esterolytic Antibodies Correlate with Structural Diversity Authors: Zemel, R. / Schindler, D.G. / Tawfik, D.S. / Eshhar, Z. / Green, B.S. #2: Journal: Structure / Year: 1994Title: Crystal Structure of a Catalytic Antibody Fab with Esterase-Like Activity Authors: Golinelli-Pimpaneau, B. / Gigant, B. / Bizebard, T. / Navaza, J. / Saludjian, P. / Zemel, R. / Tawfik, D.S. / Eshhar, Z. / Green, B.S. / Knossow, M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1kno.cif.gz | 233.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1kno.ent.gz | 187.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1kno.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/kn/1knoftp://data.pdbj.org/pub/pdb/validation_reports/kn/1kno | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|




















-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||
| 2 | 
| ||||||||||||
| 3 | 
| ||||||||||||
| Unit cell |
| ||||||||||||
| Atom site foot note | 1: CIS PROLINE - PRO A 8 / 2: CIS PROLINE - PRO A 141 / 3: CIS PROLINE - PRO B 157 / 4: CIS PROLINE - PRO B 159 / 5: CIS PROLINE - PRO B 208 / 6: CIS PROLINE - PRO C 8 / 7: CIS PROLINE - PRO C 141 / 8: CIS PROLINE - PRO D 157 / 9: CIS PROLINE - PRO D 159 / 10: CIS PROLINE - PRO D 208 / 11: CIS PROLINE - PRO E 8 / 12: CIS PROLINE - PRO E 141 / 13: CIS PROLINE - PRO F 157 / 14: CIS PROLINE - PRO F 159 / 15: CIS PROLINE - PRO F 208 | ||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper:
| ||||||||||||
| Details | MTRIX THE TRANSFORMATIONS PRESENTED ON MTRIX RECORDS BELOW DESCRIBE NON-CRYSTALLOGRAPHIC RELATIONSHIPS AMONG THE VARIOUS DOMAINS IN THIS ENTRY. APPLYING THE APPROPRIATE MTRIX TRANSFORMATION TO THE RESIDUES LISTED FIRST WILL YIELD APPROXIMATE COORDINATES FOR THE RESIDUES LISTED SECOND. APPLIED TO TRANSFORMED TO MTRIX RESIDUES RESIDUES RMSD M1 C 1 .. D 235 A 1 .. B 235 0.507 M2 E 1 .. F 235 A 1 .. B 235 0.453 |
-Components
| #1: Antibody | Mass: 23574.941 Da / Num. of mol.: 3 / Source method: isolated from a natural source / Source: (natural) #2: Antibody | Mass: 23426.248 Da / Num. of mol.: 3 / Source method: isolated from a natural source / Source: (natural) #3: Chemical | ChemComp-ZN / |   Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn#4: Chemical | 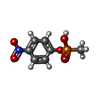  Mass: 217.116 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C7H8NO5P Mass: 217.116 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C7H8NO5PHas protein modification | Y | Sequence details | RESIDUE NUMBERING FOLLOWS THE ORDER OF APPEARANCE IN THE SEQUENCE. THIS NUMBERING CORRESPONDS TO ...RESIDUE NUMBERING FOLLOWS THE ORDER OF APPEARANCE | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.19 Å3/Da / Density % sol: 61.45 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 8 / Details: pH 8.0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal | *PLUS Density % sol: 55 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 277 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: LURE  / Beamline: DW32 / Wavelength: 0.901 Å / Beamline: DW32 / Wavelength: 0.901 Å |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Nov 23, 1994 / Details: BENT MIRROR |
| Radiation | Monochromator: GRAPHITE / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.901 Å / Relative weight: 1 |
| Reflection | Resolution: 3.2→30 Å / Num. obs: 27401 / % possible obs: 96 % / Observed criterion σ(I): 1 / Redundancy: 1.8 % / Rmerge(I) obs: 0.112 |
| Reflection | *PLUS Rmerge(I) obs: 0.112 |
| Reflection shell | *PLUS Highest resolution: 3.2 Å / Lowest resolution: 3.3 Å / % possible obs: 89 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 3.2→7 Å / σ(F): 2 Details: RESIDUES 212 - 214 OF THE LIGHT CHAINS AND 136 - 144, 234 - 235 OF THE HEAVY CHAINS ARE POORLY DEFINED BY THE ELECTRON DENSITY. CARE SHOULD ALSO BE EXERCISED IN INTERPRETING THIS MODEL, DUE ...Details: RESIDUES 212 - 214 OF THE LIGHT CHAINS AND 136 - 144, 234 - 235 OF THE HEAVY CHAINS ARE POORLY DEFINED BY THE ELECTRON DENSITY. CARE SHOULD ALSO BE EXERCISED IN INTERPRETING THIS MODEL, DUE TO THE LIMITED RESOLUTION.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 34.2 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.35 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.2→7 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
