 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1kms: HUMAN DIHYDROFOLATE REDUCTASE COMPLEXED WITH NADPH AND 6-([5-QUIN... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1kms | ||||||
|---|---|---|---|---|---|---|---|
| Title | HUMAN DIHYDROFOLATE REDUCTASE COMPLEXED WITH NADPH AND 6-([5-QUINOLYLAMINO]METHYL)-2,4-DIAMINO-5-METHYLPYRIDO[2,3-D]PYRIMIDINE (SRI-9439), A LIPOPHILIC ANTIFOLATE | ||||||
 Components Components | DIHYDROFOLATE REDUCTASE | ||||||
 Keywords Keywords | OXIDOREDUCTASE / ANTIPARASITIC DRUGS / REDUCTASE / LIPOPHILIC ANTIFOLATES | ||||||
| Function / homology |  Function and homology information Function and homology informationregulation of removal of superoxide radicals / tetrahydrobiopterin biosynthetic process / Metabolism of folate and pterines / tetrahydrofolate metabolic process / response to methotrexate / sequence-specific mRNA binding / folic acid binding / axon regeneration / dihydrofolate metabolic process / dihydrofolate reductase ...regulation of removal of superoxide radicals / tetrahydrobiopterin biosynthetic process / Metabolism of folate and pterines / tetrahydrofolate metabolic process / response to methotrexate / sequence-specific mRNA binding / folic acid binding / axon regeneration / dihydrofolate metabolic process / dihydrofolate reductase / G1/S-Specific Transcription / dihydrofolate reductase activity / folic acid metabolic process / tetrahydrofolate biosynthetic process / NADPH binding / 'de novo' pyrimidine nucleobase biosynthetic process / one-carbon metabolic process / Tetrahydrobiopterin (BH4) synthesis, recycling, salvage and regulation / mRNA regulatory element binding translation repressor activity / replication fork / NADP binding / DNA replication / negative regulation of translation / mRNA binding / mitochondrion / nucleus / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.09 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.09 Å | ||||||
 Authors Authors | Klon, A.E. / Heroux, A. / Ross, L.J. / Pathak, V. / Johnson, C.A. / Piper, J.R. / Borhani, D.W. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2002 Title: Atomic structures of human dihydrofolate reductase complexed with NADPH and two lipophilic antifolates at 1.09 a and 1.05 a resolution. Authors: Klon, A.E. / Heroux, A. / Ross, L.J. / Pathak, V. / Johnson, C.A. / Piper, J.R. / Borhani, D.W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1kms.cif.gz | 116.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1kms.ent.gz | 90.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1kms.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/km/1kmsftp://data.pdbj.org/pub/pdb/validation_reports/km/1kms | HTTPS FTP |
|---|
-Related structure data










-Links
 PDBj
PDBj






- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
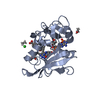
| #1: Protein | Mass: 21349.525 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Plasmid: PDFR / Species (production host): Escherichia coli / Cellular location (production host): CYTOPLASM / Production host:  | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical |   Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4#3: Chemical | ChemComp-LIH / | 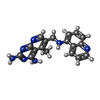  Mass: 331.374 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H17N7 Mass: 331.374 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H17N7#4: Chemical | ChemComp-NDP / | 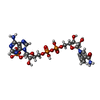  Mass: 745.421 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H30N7O17P3 Mass: 745.421 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H30N7O17P3#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 399 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 399 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.23 Å3/Da / Density % sol: 46 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 8 Details: THE TERNARY COMPLEX OF DHFR WITH NADPH AND SRI-9439 WAS FORMED BY MIXING HUMAN DHFR (20 MG/ML IN 25 MM KPO4 (PH 7.0), 0.1 MM EDTA, AND 3 MM NAN3) WITH 60 MM NADPH, FOLLOWED 15 MINUTES LATER ...Details: THE TERNARY COMPLEX OF DHFR WITH NADPH AND SRI-9439 WAS FORMED BY MIXING HUMAN DHFR (20 MG/ML IN 25 MM KPO4 (PH 7.0), 0.1 MM EDTA, AND 3 MM NAN3) WITH 60 MM NADPH, FOLLOWED 15 MINUTES LATER BY 60 MM OF INHIBITOR IN DMSO (FINAL CONCENTRATIONS OF 2 MM NADPH AND 2 MM INHIBITOR). THIS COMPLEX SOLUTION WAS MIXED WITH AN EQUAL VOLUME OF PRECIPITANT CONTAINING 24-33% PEG 4000, 0.2 M LI2SO4, 0.1 M TRIS CL (PH 7.9-8.4), AND EQUILIBRATED WITH THE PRECIPITANT BY HANGING DROP VAPOR DIFFUSION AT 277 K. THE CRYSTAL GREW IN ABOUT 3 WEEKS. THE CRYSTAL WAS FLASH-COOLED DIRECTLY IN LIQUID NITROGEN AFTER HARVESTING INTO MOTHER LIQUOR CONTAINING 10% GLYCEROL., pH 8.00, VAPOR DIFFUSION, HANGING DROP | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / pH: 7 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL  / Beamline: BL7-1 / Wavelength: 1.08 / Beamline: BL7-1 / Wavelength: 1.08 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Jun 25, 1997 / Details: PT-COATED MIRROR |
| Radiation | Monochromator: SI (111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.08 Å / Relative weight: 1 |
| Reflection | Resolution: 1.09→20 Å / Num. all: 79611 / Num. obs: 79611 / % possible obs: 99.6 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 3.6 % / Biso Wilson estimate: 10.8 Å2 / Rmerge(I) obs: 0.047 / Rsym value: 0.047 / Net I/σ(I): 9.4 |
| Reflection shell | Resolution: 1.09→1.15 Å / Redundancy: 3.3 % / Rmerge(I) obs: 0.422 / Mean I/σ(I) obs: 2.3 / Rsym value: 0.422 / % possible all: 99.1 |
| Reflection | *PLUS Highest resolution: 1.09 Å / Lowest resolution: 20 Å / Num. measured all: 289350 / Rmerge(I) obs: 0.047 |
| Reflection shell | *PLUS % possible obs: 99.1 % / Rmerge(I) obs: 0.422 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: UNPUBLISHED HUMAN DHFR STRUCTURE. Resolution: 1.09→20 Å / Num. parameters: 18984 / Num. restraintsaints: 23468 / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / Stereochemistry target values: ENGH & HUBER Details: REFINEMENT IN X-PLOR, ALTERNATED WITH MANUAL REBUILDING IN O, RESULTING IN A FREE R-FACTOR OF 24.0%. SRI-9439 WAS THEN ADDED TO THE MODEL IN THE ACTIVE SITE AND THE CIS PEPTIDE BONDS BETWEEN ...Details: REFINEMENT IN X-PLOR, ALTERNATED WITH MANUAL REBUILDING IN O, RESULTING IN A FREE R-FACTOR OF 24.0%. SRI-9439 WAS THEN ADDED TO THE MODEL IN THE ACTIVE SITE AND THE CIS PEPTIDE BONDS BETWEEN RESIDUES ARG-65 AND PRO-66 AND RESIDUES GLY-116 AND GLY-117 BECAME APPARENT IN 2FO-FC AND FO-FC MAPS. REFINEMENT PROCEEDED WITH THE ADDITION OF RIDING HYDROGEN ATOMS AND ANISOTROPIC TEMPERATURE FACTORS IN REFMAC AND ARP, YIELDING A FREE R-FACTOR OF 19.4%. AT THIS POINT, MOST OF THE SIDE CHAIN ALTERNATE CONFORMATIONS WERE MODELED. ANISOTROPIC DISPLACEMENT PARAMETERS WERE REFINED IN SHELXL AND RIDING HYDROGEN ATOMS WERE ADDED. IN THE FINAL ROUNDS OF REFINEMENT, RESTRAINTS FOR ALL ATOMS IN SRI-9439 EXCEPT FOR PLANARITY RESTRAINTS ON THE 5-QUINOLYLAMINO GROUP, AND ALL NADPH ATOMS EXCEPT FOR THOSE IN THE ADENINE RING AND ADENINE RIBOSE-2-PHOSPHATE WERE REMOVED, GIVING THE FINAL FREE R-FACTOR (17.3%). ATTEMPTS TO RESTRAIN THE PLANARITY OF THE 5-METHYL-5-DEAZAPTERIDINE MOIETY OF SRI-9439 RESULTED IN LARGE DIFFERENCE ELECTRON DENSITY PEAKS FOR THE EXOCYCLIC N4 AND C5A ATOMS, CONFIRMING THEIR OUT-OF-PLANE POSITIONS.
| |||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: MOEWS & KRETSINGER, J.MOL.BIOL.91(1973)201-228 | |||||||||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 66 / Occupancy sum hydrogen: 1406.12 / Occupancy sum non hydrogen: 1927.22 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.09→20 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||
| Software | *PLUS Name: SHELXL / Version: 97 / Classification: refinement | |||||||||||||||||||||||||||||||||
| Refinement | *PLUS % reflection Rfree: 5 % / Rfactor Rfree: 0.173 / Rfactor Rwork: 0.131 | |||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
